Package Insert
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Snake Bite Protocol
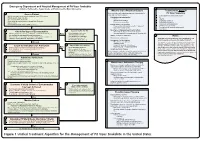
Lavonas et al. BMC Emergency Medicine 2011, 11:2 Page 4 of 15 http://www.biomedcentral.com/1471-227X/11/2 and other Rocky Mountain Poison and Drug Center treatment of patients bitten by coral snakes (family Ela- staff. The antivenom manufacturer provided funding pidae), nor by snakes that are not indigenous to the US. support. Sponsor representatives were not present dur- At the time this algorithm was developed, the only ing the webinar or panel discussions. Sponsor represen- antivenom commercially available for the treatment of tatives reviewed the final manuscript before publication pit viper envenomation in the US is Crotalidae Polyva- ® for the sole purpose of identifying proprietary informa- lent Immune Fab (ovine) (CroFab , Protherics, Nash- tion. No modifications of the manuscript were requested ville, TN). All treatment recommendations and dosing by the manufacturer. apply to this antivenom. This algorithm does not con- sider treatment with whole IgG antivenom (Antivenin Results (Crotalidae) Polyvalent, equine origin (Wyeth-Ayerst, Final unified treatment algorithm Marietta, Pennsylvania, USA)), because production of The unified treatment algorithm is shown in Figure 1. that antivenom has been discontinued and all extant The final version was endorsed unanimously. Specific lots have expired. This antivenom also does not consider considerations endorsed by the panelists are as follows: treatment with other antivenom products under devel- opment. Because the panel members are all hospital- Role of the unified treatment algorithm -

COTTONMOUTH Agkistrodon Piscivorus
COTTONMOUTH Agkistrodon piscivorus Agkistrodon is derived from ankistron and odon which in Greek mean “fishhook” and “tooth or teeth;” referring to the curved fangs of this species. Piscivorus is derived from piscis and voro which in Latin mean “fish” and “to eat”. Another common name for cottonmouth is water moccasin. The Cottonmouth is venomous. While its bite is rarely fatal, tissue damage is likely to occur and can be severe if not treated promptly. IDENTIFICATION Appearance: The cottonmouth is a stout- bodied venomous snake that reaches lengths of 30 to 42 inches as adults. Most adults are uniformly dark brown, olive, or black, tending to lose the cross banded patterning with age. Some individuals may have a dark cheek stripe (upper right image). The cottonmouth has the diagnostic features of the pit-viper family such as a wedge-shaped head, sensory pits between the eyes and nostrils, and vertical “cat-like” pupils. Juveniles are lighter and more boldly patterned with a yellow coloration toward the tip of the tail (lower right image). Dorsal scales are weakly keeled, and the subcaudal scales form only one row. Cottonmouths also have a single anal Mike Redmer plate. Subspecies: There are three subspecies of the cottonmouth. The Western Cottonmouth (A. p. leucostoma) is the only subspecies found in the Midwest. The term leucostoma refers to the white interior of mouth. Confusing Species: The non-venomous watersnakes (Nerodia) are commonly confused with Cottonmouths across their range, simply because they are snakes in water. Thus it is important to note that Cottonmouths are only found in southernmost Midwest. -

Pit Vipers: from Fang to Needle—Three Critical Concepts for Clinicians
Tuesday, July 28, 2021 Pit Vipers: From Fang to Needle—Three Critical Concepts for Clinicians Keith J. Boesen, PharmD & Nicholas B. Hurst, M.D., MS Disclosures / Potential Conflicts of Interest • Keith Boesen and Nicholas Hurst are employed by Rare Disease Therapeutics, Inc. (RDT) • RDT is a U.S. company working with Laboratorios Silanes, S.A. de C.V., a company in Mexico • Laboratorios Silanes manufactures a variety of antivenoms Note: This program may contain the mention of suppliers, brands, products, services or drugs presented in a case study or comparative format using evidence-based research. Such examples are intended for educational and informational purposes and should not be perceived as an endorsement of any particular supplier, brand, product, service or drug. 2 Learning Objectives At the end of this session, participants should be able to: 1. Describe the venom variability in North American Pit Vipers 2. Evaluate the clinical symptoms associated with a North American Pit Viper envenomation 3. Develop a treatment plan for a North American Pit Viper envenomation 3 Audience Poll Question: #1 of 5 My level of expertise in treating Pit Viper Envenomation is… a. I wouldn’t know where to begin! b. I have seen a few cases… c. I know a thing or two because I’ve seen a thing or two d. I frequently treat these patients e. When it comes to Pit Viper envenomation, I am a Ssssuper Sssskilled Ssssnakebite Sssspecialist!!! 4 PIT VIPER ENVENOMATIONS PIT VIPERS Loreal Pits Movable Fangs 1. Russel 1983 -Photo provided by the Arizona Poison and Drug Information Center 1. -

Long-Term Effects of Snake Envenoming
toxins Review Long-Term Effects of Snake Envenoming Subodha Waiddyanatha 1,2, Anjana Silva 1,2 , Sisira Siribaddana 1 and Geoffrey K. Isbister 2,3,* 1 Faculty of Medicine and Allied Sciences, Rajarata University of Sri Lanka, Saliyapura 50008, Sri Lanka; [email protected] (S.W.); [email protected] (A.S.); [email protected] (S.S.) 2 South Asian Clinical Toxicology Research Collaboration, Faculty of Medicine, University of Peradeniya, Peradeniya 20400, Sri Lanka 3 Clinical Toxicology Research Group, University of Newcastle, Callaghan, NSW 2308, Australia * Correspondence: [email protected] or [email protected]; Tel.: +612-4921-1211 Received: 14 March 2019; Accepted: 29 March 2019; Published: 31 March 2019 Abstract: Long-term effects of envenoming compromise the quality of life of the survivors of snakebite. We searched MEDLINE (from 1946) and EMBASE (from 1947) until October 2018 for clinical literature on the long-term effects of snake envenoming using different combinations of search terms. We classified conditions that last or appear more than six weeks following envenoming as long term or delayed effects of envenoming. Of 257 records identified, 51 articles describe the long-term effects of snake envenoming and were reviewed. Disability due to amputations, deformities, contracture formation, and chronic ulceration, rarely with malignant change, have resulted from local necrosis due to bites mainly from African and Asian cobras, and Central and South American Pit-vipers. Progression of acute kidney injury into chronic renal failure in Russell’s viper bites has been reported in several studies from India and Sri Lanka. Neuromuscular toxicity does not appear to result in long-term effects. -

Snakebite: the World's Biggest Hidden Health Crisis
Snakebite: The world's biggest hidden health crisis Snakebite is a potentially life-threatening neglected tropical disease (NTD) that is responsible for immense suffering among some 5.8 billion people who are at risk of encountering a venomous snake. The human cost of snakebite Snakebite Treatment Timeline Each year, approximately 5.4 million people are bitten by a snake, of whom 2.7 million are injected with venom. The first snake antivenom This leads to 400,000 people being permanently dis- produced, against the Indian Cobra. abled and between 83,000-138,000 deaths annually, Immunotherapy with animal- mostly in sub-Saharan Africa and South Asia. 1895 derived antivenom has continued to be the main treatment for snakebite evenoming for 120 years Snakebite: both a consequence and a cause of tropical poverty The Fav-Afrique antivenom, 2014 produced by Sanofi Pasteur (France) Survivors of untreated envenoming may be left with permanently discontinued amputation, blindness, mental health issues, and other forms of disability that severely affect their productivity. World Health Organization Most victims are agricultural workers and children in 2018 (WHO) lists snakebite envenoming the poorest parts of Africa and Asia. The economic as a neglected tropical disease cost of treating snakebite envenoming is unimaginable in most communities and puts families and communi- ties at risk of economic peril just to pay for treatment. WHO launches a strategy to prevent and control snakebite envenoming, including a program targeting affected communities and their health systems Global antivenom crisis 2019 The world produces less than half of the antivenom it The Scientific Research Partnership needs, and this only covers 57% of the world’s species for Neglected Tropical Snakbites of venomous snake. -

Bitis Arietans) Venom, and Their Neutralization by Antivenom
Toxins 2014, 6, 1586-1597; doi:10.3390/toxins6051586 OPEN ACCESS toxins ISSN 2072-6651 www.mdpi.com/journal/toxins Article In Vitro Toxic Effects of Puff Adder (Bitis arietans) Venom, and Their Neutralization by Antivenom Steven Fernandez 1, Wayne Hodgson 1, Janeyuth Chaisakul 2, Rachelle Kornhauser 1, Nicki Konstantakopoulos 1, Alexander Ian Smith 3 and Sanjaya Kuruppu 3,* 1 Department of Pharmacology, Monash University, Building 13E, Wellington Road, Clayton, Vic 3800, Australia; E-Mails: [email protected] (S.F.); [email protected] (W.H.); [email protected] (R.K.); [email protected] (N.K.) 2 Department of Pharmacology, Phramongkutklao College of Medicine, Bangkok 10400, Thailand; E-Mail: [email protected] 3 Department of Biochemistry & Molecular Biology, Monash University, Building 77, Wellington Road, Clayton, Vic 3800, Australia; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +61-3-9902-9372; Fax: + 61-3-9902-9500. Received: 24 November 2013; in revised form: 6 April 2014 / Accepted: 4 May 2014 / Published: 19 May 2014 Abstract: This study investigated the in vitro toxic effects of Bitis arietans venom and the ability of antivenom produced by the South African Institute of Medical Research (SAIMR) to neutralize these effects. The venom (50 µg/mL) reduced nerve-mediated twitches of the chick biventer muscle to 19% ± 2% of initial magnitude (n = 4) within 2 h. This inhibitory effect of the venom was significantly attenuated by prior incubation of tissues with SAIMR antivenom (0.864 µg/µL; 67% ± 4%; P < 0.05; n = 3–5, unpaired t-test). -

Medicines/Pharmaceuticals of Animal Origin V3.0 November 2020
Medicines/pharmaceuticals of animal origin V3.0 November 2020 Medicines/pharmaceuticals of animal origin - This guideline provides information for all clinical staff within Hospital and Health Services (HHS) on best practice for avoidance of issues related to animal products. Medicines/pharmaceuticals of animal origin - V3.0 November 2020 Published by the State of Queensland (Queensland Health), November 2020 This document is licensed under a Creative Commons Attribution 3.0 Australia licence. To view a copy of this licence, visit creativecommons.org/licenses/by/3.0/au © State of Queensland (Queensland Health) 2020 You are free to copy, communicate and adapt the work, as long as you attribute the State of Queensland (Queensland Health). For more information contact: Medication Services Queensland, Queensland Health, GPO Box 48, Brisbane QLD 4001, email [email protected] An electronic version of this document is available at https://www.health.qld.gov.au/__data/assets/pdf_file/0024/147507/qh-gdl-954.pdf Disclaimer: The content presented in this publication is distributed by the Queensland Government as an information source only. The State of Queensland makes no statements, representations or warranties about the accuracy, completeness or reliability of any information contained in this publication. The State of Queensland disclaims all responsibility and all liability (including without limitation for liability in negligence) for all expenses, losses, damages and costs you might incur as a result of the information being inaccurate -

Epidemiology and Clinical Outcomes of Snakebite in the Elderly: a Toxic Database Study
Clinical Toxicology ISSN: 1556-3650 (Print) 1556-9519 (Online) Journal homepage: http://www.tandfonline.com/loi/ictx20 Epidemiology and clinical outcomes of snakebite in the elderly: a ToxIC database study Meghan B. Spyres, Anne-Michelle Ruha, Kurt Kleinschmidt, Rais Vohra, Eric Smith & Angela Padilla-Jones To cite this article: Meghan B. Spyres, Anne-Michelle Ruha, Kurt Kleinschmidt, Rais Vohra, Eric Smith & Angela Padilla-Jones (2018) Epidemiology and clinical outcomes of snakebite in the elderly: a ToxIC database study, Clinical Toxicology, 56:2, 108-112, DOI: 10.1080/15563650.2017.1342829 To link to this article: https://doi.org/10.1080/15563650.2017.1342829 Published online: 13 Jul 2017. Submit your article to this journal Article views: 120 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=ictx20 CLINICAL TOXICOLOGY, 2018 VOL. 56, NO. 2, 108–112 https://doi.org/10.1080/15563650.2017.1342829 CLINICAL RESEARCH Epidemiology and clinical outcomes of snakebite in the elderly: a ToxIC Ã database study Meghan B. Spyresa, Anne-Michelle Ruhab, Kurt Kleinschmidtc, Rais Vohrad, Eric Smithc and Angela Padilla-Jonesb aDepartment of Emergency Medicine, Division of Medical Toxicology, University of Southern California, Los Angeles, CA, USA; bDepartment of Medical Toxicology, Banner-University Medical Center Phoenix, Phoenix, AZ, USA; cDepartment of Emergency Medicine, Univesity of Texas Southwestern Medical Center, Dallas, TX, USA; dDepartment of Emergency Medicine, UCSF Fresno Medical Center, Fresno, CA, USA ABSTRACT ARTICLE HISTORY Introduction: Epidemiologic studies of snakebites in the United States report typical victims to be Received 23 February 2017 young men. -

Reproductive Biology and Natural History of the White-Lipped Pit Viper (Trimeresurus Albolabris Gray, 1842) in Hong Kong Anne Devan-Song University of Rhode Island
University of Rhode Island DigitalCommons@URI Natural Resources Science Faculty Publications Natural Resources Science 2017 Reproductive Biology and Natural History of the White-lipped Pit Viper (Trimeresurus albolabris Gray, 1842) in Hong Kong Anne Devan-Song University of Rhode Island Paolo Martelli See next page for additional authors Follow this and additional works at: https://digitalcommons.uri.edu/nrs_facpubs Citation/Publisher Attribution Devan-Song, A., Martelli, P., & Karraker, N. E. (2017). Reproductive Biology and Natural History of the White-lipped Pit Viper (Trimeresurus albolabris Gray, 1842) in Hong Kong. Herpetological Conservation and Biology, 12(1), 41-55. Retrieved from http://www.herpconbio.org/Volume_12/Issue_1/Devan-Song_etal_2017.pdf Available at: http://www.herpconbio.org/Volume_12/Issue_1/Devan-Song_etal_2017.pdf This Article is brought to you for free and open access by the Natural Resources Science at DigitalCommons@URI. It has been accepted for inclusion in Natural Resources Science Faculty Publications by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. Authors Anne Devan-Song, Paolo Martelli, and Nancy E. Karraker This article is available at DigitalCommons@URI: https://digitalcommons.uri.edu/nrs_facpubs/115 Herpetological Conservation and Biology 12:41–55. Submitted: 30 September 2015; Accepted: 18 January 2017; Published: 30 April 2017. Reproductive Biology and Natural History of the White-lipped Pit Viper (Trimeresurus albolabris Gray, -

Movement and Home Range of Green Pit Vipers (Trimeresurus Spp.) in a Rural Landscape in North-East Thailand
RESEARCH ARTICLE The Herpetological Bulletin 142, 2017: 19-28 Movement and home range of green pit vipers (Trimeresurus spp.) in a rural landscape in north-east Thailand CURT H. BARNES1, COLIN T. STRINE1*, PONGTHEP SUWANWAREE1, & JACQUES G. HILL III2 1School of Biology, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, Thailand 2Department of Biological Science, University of Arkansas, Fayetteville, AR, USA *Corresponding author Email: [email protected] ABSTRACT - Rural communities and agricultural landscapes serve as important areas for biodiversity, yet study of snakes in these fragmented environments is severely lacking. Hospital records indicate that green pit vipers (Trimeresurus spp.) inflict the highest number of venomous snakebites of any snake group in the Nakhon Ratchasima, Pak Thong Chai, and Wang Nam Khieo rural regions of north-east Thailand, causing debilitating injuries and, subsequently, negative perceptions of these species. We utilised radio telemetry to assess male and female Trimeresurus albolabris (N = 1, N = 1) and T. macrops (N = 2, N = 7) movements and home ranges in rural portions of the the Sakaerat Biosphere Reserve in north-east Thailand from October 2015 through January 2017. Green pit vipers of both species were tracked for a mean of 97.6 ± 15 days (median = 88, range 35-190). They moved a mean distance of 26.3 ± 3.32 meters between locations (median = 25.11, range 13.2-50.4) and exhibited mean minimum convex polygon home ranges of 0.14 ± 0.043 hectares (median = 0.095, range 0.006-0.423). Big-eyed green pit vipers (T. macrops) differed in movement patterns and home range size by sex and fecundity, although not statistically so. -

Polyvalent Horse F(Ab`)2 Snake Antivenom: Development of Process to Produce Polyvalent Horse F(Ab`)2 Antibodies Anti-African Snake Venom
African Journal of Biotechnology Vol. 9 (16), pp. 2446-2455, 19 April, 2010 Available online at http://www.academicjournals.org/AJB ISSN 1684–5315 © 2009 Academic Journals Full Length Research Paper Polyvalent horse F(Ab`)2 snake antivenom: Development of process to produce polyvalent horse F(Ab`)2 antibodies anti-african snake venom R. G. Guidlolin1, R. M. Marcelino1, H. H. Gondo1, J. F. Morais1, R. A. Ferreira2, C. L. Silva2, T. L. Kipnis2, J. A. Silva2, J. Fafetine3 and W. D. da Silva2* 1Divisao Bioindustrial, Instituto Butantan, Sao Paulo, SP, Brazil. 2Laboratório de Biologia do Reconhecer, Centro de Biociencias e Biotecnologia, Universidade Estadual do Norte Fluminense – Darcy Ribeiro, Campos dos Goytacazes, RJ, Brazil. 3Universidade Eduardo Mondlane, Maputo, Mozambique. Accepted 23 March, 2010 A method to obtain polyvalent anti-Bitis and polyvalent-anti-Naja antibodies was developed by immunizing horses with B. arietans, B. nasicornis, B. rhinoceros, N. melanoleuca and N. mossambica crude venoms. Antibody production was followed by the ELISA method during the immunization procedure. Once the desired anti-venom antibody titers were attained, horses were bled and the immunoglobulins were separated from the sera by (NH4)2SO4 precipitation, cleaved with pepsin and filtered through a 30 kDa ultrafiltration membrane. F(ab´)2 fragments were further purified by Q-Fast Flow chromatography, concentrated by molecular ultrafiltration and sterilized by filtration through 0.22 m membranes. The resulting F(ab´)2 preparations were rich in intact L and in pieces of H IgG(T) chains, as demonstrated by electrophoresis and Western blot and exhibited high antibody titers, as assayed by the ELISA method. -

Venomous Nonvenomous Snakes of Florida
Venomous and nonvenomous Snakes of Florida PHOTOGRAPHS BY KEVIN ENGE Top to bottom: Black swamp snake; Eastern garter snake; Eastern mud snake; Eastern kingsnake Florida is home to more snakes than any other state in the Southeast – 44 native species and three nonnative species. Since only six species are venomous, and two of those reside only in the northern part of the state, any snake you encounter will most likely be nonvenomous. Florida Fish and Wildlife Conservation Commission MyFWC.com Florida has an abundance of wildlife, Snakes flick their forked tongues to “taste” their surroundings. The tongue of this yellow rat snake including a wide variety of reptiles. takes particles from the air into the Jacobson’s This state has more snakes than organs in the roof of its mouth for identification. any other state in the Southeast – 44 native species and three nonnative species. They are found in every Fhabitat from coastal mangroves and salt marshes to freshwater wetlands and dry uplands. Some species even thrive in residential areas. Anyone in Florida might see a snake wherever they live or travel. Many people are frightened of or repulsed by snakes because of super- stition or folklore. In reality, snakes play an interesting and vital role K in Florida’s complex ecology. Many ENNETH L. species help reduce the populations of rodents and other pests. K Since only six of Florida’s resident RYSKO snake species are venomous and two of them reside only in the northern and reflective and are frequently iri- part of the state, any snake you en- descent.