Electron and Ion Spectroscopy of Metal Hydrocarbon Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Planar Cyclopenten‐4‐Yl Cations: Highly Delocalized Π Aromatics
Angewandte Research Articles Chemie How to cite: Angew.Chem. Int. Ed. 2020, 59,18809–18815 Carbocations International Edition: doi.org/10.1002/anie.202009644 German Edition: doi.org/10.1002/ange.202009644 Planar Cyclopenten-4-yl Cations:Highly Delocalized p Aromatics Stabilized by Hyperconjugation Samuel Nees,Thomas Kupfer,Alexander Hofmann, and Holger Braunschweig* 1 B Abstract: Theoretical studies predicted the planar cyclopenten- being energetically favored by 18.8 kcalmolÀ over 1 (MP3/ 4-yl cation to be aclassical carbocation, and the highest-energy 6-31G**).[11–13] Thebishomoaromatic structure 1B itself is + 1 isomer of C5H7 .Hence,its existence has not been verified about 6–14 kcalmolÀ lower in energy (depending on the level experimentally so far.Wewere now able to isolate two stable of theory) than the classical planar structure 1C,making the derivatives of the cyclopenten-4-yl cation by reaction of bulky cyclopenten-4-yl cation (1C)the least favorable isomer.Early R alanes Cp AlBr2 with AlBr3.Elucidation of their (electronic) solvolysis studies are consistent with these findings,with structures by X-raydiffraction and quantum chemistry studies allylic 1A being the only observable isomer, notwithstanding revealed planar geometries and strong hyperconjugation the nature of the studied cyclopenteneprecursor.[14–18] Thus, interactions primarily from the C Al s bonds to the empty p attempts to generate isomer 1C,orits homoaromatic analog À orbital of the cationic sp2 carbon center.Aclose inspection of 1B,bysolvolysis of 4-Br/OTs-cyclopentene -

An Analysis of Electrophilic Aromatic Substitution: a “Complex Approach” PCCP
Volume 23 Number 9 7 March 2021 Pages 5033–5682 PCCP Physical Chemistry Chemical Physics rsc.li/pccp ISSN 1463-9076 PERSPECTIVE Janez Cerkovnik et al . An analysis of electrophilic aromatic substitution: a “complex approach” PCCP View Article Online PERSPECTIVE View Journal | View Issue An analysis of electrophilic aromatic substitution: a ‘‘complex approach’’† Cite this: Phys. Chem. Chem. Phys., a a b 2021, 23, 5051 Nikola Stamenkovic´, Natasˇa Poklar Ulrih and Janez Cerkovnik * Electrophilic aromatic substitution (EAS) is one of the most widely researched transforms in synthetic organic chemistry. Numerous studies have been carried out to provide an understanding of the nature of its reactivity pattern. There is now a need for a concise and general, but detailed and up-to-date, overview. The basic principles behind EAS are essential to our understanding of what the mechanisms underlying EAS are. To date, textbook overviews of EAS have provided little information about the Received 5th October 2020, mechanistic pathways and chemical species involved. In this review, the aim is to gather and present the Accepted 21st December 2020 up-to-date information relating to reactivity in EAS, with the implication that some of the key concepts DOI: 10.1039/d0cp05245k will be discussed in a scientifically concise manner. In addition, the information presented herein suggests certain new possibilities to advance EAS theory, with particular emphasis on the role of modern Creative Commons Attribution-NonCommercial 3.0 Unported Licence. rsc.li/pccp -

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -

Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2
Metal-Ligand Bonding and Inorganic Reaction Mechanisms Year 2 RED Metal-ligand and metal-metal bonding of the transition metal elements Synopsis Lecture 1: Trends of the transition metal series. Ionic vs Covalent bonding. Nomenclature. Electron counting. Lecture 2: Thermodynamics of complex formation. Why complexes form. Recap of molecular orbital theory. 18-electron rule. Lecture 3: Ligand classes. -donor complexes. Octahedral ML6 molecular orbital energy diagram. Lecture 3: - acceptor ligands and synergic bonding. Binding of CO, CN , N2, O2 and NO. Lecture 4: Alkenes, M(H2) vs M(H)2, Mn(O2) complexes, PR3. Lecture 5: 2- - 2- 3- donor ligands, metal-ligand multiple bonds, O , R2N , RN , N . Lecture 6: ML6 molecular orbital energy diagrams incorporating acceptor and donor ligands. Electron counting revisited and link to spectrochemical series. Lecture 7: Kinetics of complex formation. Substitution mechanisms of inorganic complexes. Isomerisation. Lecture 8: Ligand effects on substitution rates (trans-effect, trans-influence). Metal and geometry effects on substitution rates. Lecture 9: Outer sphere electron transfer. Lecture 10: Inner Sphere electron transfer. Bridging ligands. 2 Learning Objectives: by the end of the course you should be able to i) Use common nomenclature in transition metal chemistry. ii) Count valence electrons and determine metal oxidation state in transition metal complexes. iii) Understand the physical basis of the 18-electron rule. iv) Appreciate the synergic nature of bonding in metal carbonyl complexes. v) Understand the relationship between CO, the 'classic' -acceptor and related ligands such as NO, CN, N2, and alkenes. 2 vi) Describe the nature of the interaction between -bound diatomic molecules (H2, O2) and their relationship to -acceptor ligands. -
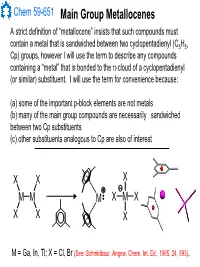
Main Group Metallocenes
Chem 59-651 Main Group Metallocenes A strict definition of “metallocene” insists that such compounds must contain a metal that is sandwiched between two cyclopentadienyl (C5H5, Cp) groups, however I will use the term to describe any compounds containing a “metal” that is bonded to the π-cloud of a cyclopentadienyl (or similar) substituent. I will use the term for convenience because: (a) some of the important p-block elements are not metals (b) many of the main group compounds are necessarily sandwiched between two Cp substituents (c) other substituents analogous to Cp are also of interest XX X MM M XMX X X X M = Ga, In, Tl; X = Cl, Br (See: Schmidbaur, Angew. Chem. Int. Ed., 1985, 24, 893). Chem 59-651 Cyclopentadienyl Ligand Basics The structural features and bonding interactions between cyclopentadienyl groups and metal atoms shows considerable variation and is described using a few different conventions (See Jutzi and Burford, Chem. Rev., 1999, 99, 969 and references therein). In most main group metallocenes, even those that are σ-bonded in the solid state, the rings rotate rapidly through a series of 1,2-shifts. A Cp group can be considered to be p-bonded to an element if the Hapticity: element sits inside the cylinder defined by the 5 carbon atoms of M MM the Cp ligand. The center of mass of the 5 C atoms is known as the centroid. η1-Cp η2-Cp η3-Cp M M M A σ-bonded Cp can be identified η4-Cp η5-Cp ≡η5-Cp by the angle at the ipso carbon M and bond lengths should indicate single bonds to the ipso carbon and a localized diene structure for σ the α and β carbon fragment. -

Application of the Covalent Bond Classification Method for The
Article pubs.acs.org/jchemeduc Application of the Covalent Bond Classification Method for the Teaching of Inorganic Chemistry Malcolm L. H. Green† and Gerard Parkin*,‡ † Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, United Kingdom ‡ Department of Chemistry, Columbia University, New York, New York 10027, United States *S Supporting Information ABSTRACT: The Covalent Bond Classification (CBC) method provides a means to classify covalent molecules according to the number and types of bonds that surround an atom of interest. This approach is based on an elementary molecular orbital analysis of the bonding involving the central atom (M), with the various interactions being classified according to the number of electrons that each neutral ligand contributes to the bonding orbital. Thus, with respect to the atom of interest (M), the ligand can contribute either two (L), one (X), or zero (Z) electrons to a bonding orbital. A normal covalent bond is represented as M−X, whereas dative covalent bonds are represented as either M←LorM→Z, according to whether the ligand is the donor (L) fi or acceptor (Z). A molecule is classi ed as [MLlXxZz] according to the number of L, X, and Z ligand functions that surround M. Not only does fi fi the [MLlXxZz] designation provide a formal classi cation of a molecule, but it also indicates the electron con guration, the valence, and the number of nonbonding electrons on M. As such, the classification allows a student to understand relationships between molecules, thereby increasing their ability to conceptualize and learn the chemistry of the elements. -

Hons.)-CHEMISTRY (Program ID-39
DAV UNIVERSITY JALANDHAR Course Scheme & Syllabus For M.Sc (Hons.)-CHEMISTRY (Program ID-39) 1st TO 4th SEMESTER 2018-2019 DAV UNIVERSITY, JALANDHAR Total minimum credits required for M.Sc. (Hons.) Chemistry are 96 Scheme of Courses M.Sc. M.Sc. (Hons.) Chemistry Semester 1 S.No Paper Code Course Type Course Title L T P Cr 1 CHE501B Core Organic Chemistry-I 4 0 0 4 2 CHE502B Core Inorganic Chemistry-I 4 0 0 4 3 CHE503B Core Physical Chemistry 4 0 0 4 4 CHE504B Core Analytical Chemistry 4 0 0 4 5 CHE505B Core Organic Chemistry Lab-I 0 0 4 2 6 CHE506B Core Inorganic Chemistry Lab-I 0 0 4 2 7 Interdisciplinary Course-I 4 Total 24 Semester 2 S.No Paper Code Course Type Course Title L T P Cr 1 CHE507B Core Organic Chemistry-II 4 0 0 4 2 CHE508B Core Inorganic Chemistry-II 4 0 0 4 3 CHE509B Core Physical Chemistry-II 4 0 0 4 4 Core CHE510B Spectroscopy-I 4 0 0 4 5 CHE511B Core Organic Chemistry Lab-II 0 0 4 2 6 CHE512B Core Physical Chemistry Lab-I 0 0 4 2 7 Interdisciplinary Course-II 4 Total 24 2 DAV UNIVERSITY, JALANDHAR Semester 3 S.No Paper Code Course Type Course Title L T P Cr 1 CHE601B Core Organic Chemistry-III 4 0 0 4 2 CHE602B Core Inorganic Chemistry-III 4 0 0 4 3 CHE603B Core Physical Chemistry-III 4 0 0 4 4 CHE604B Core Spectroscopy-II 4 0 0 4 5 CHE606B Core Inorganic Chemistry Lab-II 0 0 4 2 6 CHE607B Core Physical Chemistry Lab-II 0 0 4 2 7 CHE608B* Core Seminar and Literature Survey 0 0 0 2* 8 CHE620B* Core Advance Chemistry Lab-I 0 0 8 4* Department Elective-I 4 Total 26/28* Department Elective-I Elective 1 CHE605B Advanced Electrochemistry 4 0 0 4 2 CHE617B Elective Synthetic Organic Chemistry 4 0 0 4 Elective 3 CHE614B Molecules of Life 4 0 0 4 Elective 4 PHY670 Nanoscience and Nanotechnology 4 0 0 4 *Only 30 students in the class will be enrolled for research projects based upon their merit/performance in MSc first year. -

D. Michael P. Mingos Editor 100 Years Old and Getting Stronger
Structure and Bonding 169 Series Editor: D.M.P. Mingos D. Michael P. Mingos Editor The Chemical Bond I 100 Years Old and Getting Stronger 169 Structure and Bonding Series Editor: D.M.P. Mingos, Oxford, United Kingdom Editorial Board: F.A. Armstrong, Oxford, United Kingdom X. Duan, Beijing, China L.H. Gade, Heidelberg, Germany K.R. Poeppelmeier, Evanston, IL, USA G. Parkin, NewYork, USA M. Takano, Kyoto, Japan Aims and Scope The series Structure and Bonding publishes critical reviews on topics of research concerned with chemical structure and bonding. The scope of the series spans the entire Periodic Table and addresses structure and bonding issues associated with all of the elements. It also focuses attention on new and developing areas of modern structural and theoretical chemistry such as nanostructures, molecular electronics, designed molecular solids, surfaces, metal clusters and supramolecular structures. Physical and spectroscopic techniques used to determine, examine and model structures fall within the purview of Structure and Bonding to the extent that the focus is on the scientific results obtained and not on specialist information concerning the techniques themselves. Issues associated with the development of bonding models and generalizations that illuminate the reactivity pathways and rates of chemical processes are also relevant. The individual volumes in the series are thematic. The goal of each volume is to give the reader, whether at a university or in industry, a comprehensive overview of an area where new insights are emerging that are of interest to a larger scientific audience. Thus each review within the volume critically surveys one aspect of that topic and places it within the context of the volume as a whole. -

Organometallic Compounds
Organometallic Compounds In simpler terms these are compounds containing metal-carbon bonds Examples: CH3-MgBr, Ph-Li, [Ni(CO)4], Ferrocene etc. These compounds can be seen as having covalent bonds between the metal and the carbon atom(s). In general, compounds having a metal-ligand bond of considerable covalent character have similar chemistry and follow the chemical behavior of organometallic compounds Metal-cyano complexes are not considered as organometallic compp,ounds, while metal carbonyl complexes are. 18-electron Rule Having 18 electrons in the outer shell consisting of s, p and d orbitals is considered as an indication of stability as in inert gases. The rule suggests that compounds that can attain 18 electrons within the bonding orbitals of the metal show increased stability Mo contributes 6 electrons Benzene contributes 6 electrons Three CO contribute 6 electrons The compound follows 18-electron rule and is stable Hapticity: The number of atoms in the ligand which are directly coordinated to the metal. Hapticity is denoted as η pentahapto – η5 W contributes 6 electrons One cyclopentadiene contributes 5 electrons The other contributes 3 electrons Two CO contribute 4 electrons trihapto – η3 The compound follows 18-electron rule and is stable 18-electron Rule Metals with odd number of electrons form metal-metal bonds in their carbonyl complexes to satisfy the 18-electron rule CO OC CO CO OC Mn Mn CO OC OC CO OC Carbonyl groups can also bridge between two metals, where they can be seen as contributing one electron each to the two metals Scope of 18-electron rule for d-block organometallic compounds Usually less than Usually 18 16 or 18 18 electrons electrons Sc Ti V Cr Mn Fe Co Ni YZrNb MoTcRu RhPd La Hf Ta W Re Os Ir Pt Oxidation Numbers Oxidation numbers are important in predicting and understanding the reactivity of organometallic compounds. -

Complexes of Π −Bound Ligands Lecture 3
Module 9 : Complexes of π −bound ligands Lecture 3 : Metal cyclopentadienyl complexes Objectives In this lecture you will learn the following The cyclopentadienyl ligands. The synthesis and reactivity of metal−cyclopentadienyl complexes. The metal−cyclopentadienyl interaction. Cyclopentadienyl moiety acts as an important “spectator” ligand and is quite ubiquitous in organometallic chemistry. It remains inert to most nucleophiles and electrophiles and solely engages in stabilizing organometallic complexes. The cyclopentadienyl ligands form a wide array of organometallic compounds exhibiting different formulations that begin with the so-called “piano stool” CpMLn (n = 2,3 or 4) type ones and extends to the most commonly observed “metallocene” Cp2M type ones to even go beyond further to the “bent metallocene” Cp2MXn (n = 1,2 or 3) type ones. In the “piano stool” CpMLn structure, the cyclopentadienyl (Cp) ligand is regarded as the “seat” of the piano stool while the remaining L ligands are referred to as the “legs” of the piano stool. Though the cyclopentadienyl ligand often binds to metal in a η5 (pentahapto) fashion, e. g. as in ferrocene, the other form of binding to 3 5 3 metal at lower hapticities, like that of the η (trihapto) binding e. g. as in (η −Cp)(η −Cp)W(CO)2 and 1 5 1 that of the η (monohapto) binding e. g. as in (η −Cp)(η −Cp)Fe(CO)2, are also seen on certain rare occasions. The binding modes of the cyclopentadienyl ligand in metal complexes can be ascertained to a certain degree by 1H NMR in the diamagnetic metal complexes, in which the Cp−protons appear as a singlet between 5.5−3.5 ppm while the β and γ hydrogens come at 7−5 ppm.