An MCM Modeling Study of Nitryl Chloride (Clno2) Impacts on Oxidation, Ozone Production and Nitrogen Oxide Partitioning in Polluted Continental Outflow
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ozone Depletion, Greenhouse Gases, and Climate Change
DOCUMENT RESUME ED 324 229 SE 051 620 TITLE Ozone Depletion, Greenhouse Gaaes, and Climate Change. Proceedings of a Joint Symposium by theBoard on Atmospheric Sciences and Climate andthe Committee on Global Change, National ResearchCouncil (Washington, D.C., March 23, 1988). INSTITUTION National Academy of Sciences - National Research Council, Washington, D.C. SPONS AGENCY National Science Foundation, Washington, D.C. REPORT NO ISBN-0-309-03945-2 PUB DATE 90 NOTE 137p. AVAILABLE FROMNational Academy of Scences, National AcademyPress, 2101 Constitution Avenue, NW, Washington, DC 20418 ($20.00). PUB TYPE Collected Works Conference Proceedings (021) EDRS PRICE MF01 Plus Postage. PC Not Available from EDRS. DESCRIPTORS Air Pollution; *Climate; *Conservation(Environment); Depleted Resources; Earth Science; Ecology; *Environmental Education; *Environmental Influences; Global Approach; *Natural Resources; Science Education; Thermal Environment; World Affairs; World Problems IDENTIFIERS *Global Climate Change ABSTRACT The motivation for the organization of thissymposium was the accumulation of evidence from manysources, both short- and longterm,_that the global climate is in a state of change. Data which defy integrated explanation including temperature, ozone, methane, precipitation and other climate-related trendshave presented troubling problems for atmospheric sciencesince the 1980's. Ten papers from this symposium are presentedhere: (1) "Global Change and the Changing Atmosphere"(William C. Clark); (2) "Stratospheric Ozone Depletion: Global Processes"(Daniel L. Albritton); (3) "Stratospheric Czone Depletion: AntarcticProcesses" (Robert T. Watson); (4) "The Role of Halocarbons in Stratospheric Ozone Depletion" (F. Sherwood Rowland);(5) "Heterogenous Chemical Processes in Ozone Depletion" (Mario J. Molina);(6) "Free Radicals in the Earth's Atmosphere: Measurement andInterpretation" (James G. Anderson); (7) "Theoretical Projections of StratosphericChange Due to Increasing Greenhouse Gases and Changing OzoneConcentrations" (Jerry D. -

Significant Impact of Heterogeneous Reactions of Reactive Chlorine Species on Summertime Atmospheric Ozone and Free-Radical Form
Science of the Total Environment 693 (2019) 133580 Contents lists available at ScienceDirect Science of the Total Environment journal homepage: www.elsevier.com/locate/scitotenv Significant impact of heterogeneous reactions of reactive chlorine species on summertime atmospheric ozone and free-radical formation in north China Xionghui Qiu a,b,QiYingc,⁎, Shuxiao Wang a,b,⁎⁎, Lei Duan a,b, Yuhang Wang d,KedingLue,PengWangf, Jia Xing a,b, Mei Zheng g,MinjiangZhaoa,b, Haotian Zheng a,b, Yuanhang Zhang e,JimingHaoa,b a State Key Joint Laboratory of Environmental Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100084, China b State Environmental Protection Key Laboratory of Sources and Control of Air Pollution Complex, Beijing 100084, China c Zachry Department of Civil Engineering, Texas A&M University, College Station, TX, United States d School of Earth and Atmospheric Sciences, Georgia Institute of Technology, Atlanta, GA 30332, United States e State Key Joint Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University,Beijing,China f Department of Civil and Environmental Engineering, The Hong Kong Polytechnic University, 999077, Hong Kong, China g SKL-ESPC and BIC-ESAT, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China HIGHLIGHTS GRAPHICAL ABSTRACT • This work represents the first high reso- lution regional modeling to quantify the impact of chlorine chemistry on the ox- idation capacity in a polluted urban at- mosphere. • These heterogeneous reactions of reac- tive chlorine species increased the O3, OH, HO2 and RO2 concentrations signifi- cantly for some regions in the Beijing- Tianjin-Hebei (BTH) area. -

Nitrogen Oxychlorides : a Bibliography on Data for Physical and Chemical
NBS Publl - cations NArL INST. OF STAND & TECH NBS SPECIAL PUBLICATION 478 U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards NITROGEN OXYCHLORIDES: A Bibliography on Data for Physical and Chemical Properties of CUNO, CLNO2, and CdNOs NATIONAL BUREAU OF STANDARDS The National Bureau of Standards^ was established by an act of Congress March 3, 1901. The Bureau's overall goal is to' strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to pro- mote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute! for Applied Technology, the Institute for Computer Sciences and Technology, the Office for Information Programs, and the Office of Experimental Technology Incentives Program. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consist- ent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essen-' tial services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry,! and commerce. The Institute consists of the Office of Measurement Services, and the following center and divisions: -

New Reactions of Nitro Compounds
This dissertation has been 62-2176 microfilmed exactly as received KAPLAN, Ralph Benjamin, 1920- NEW REACTIONS OF NITRO COMPOUNDS. The Ohio State University, Ph.D., 1950 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan NEW REACTIONS OF NITRO COMPOUNDS DISSERTATION Presented In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of the Ohio State University By RALPH B. KAPLAN, B.A., The Ohio State University 1950 Approved by: Adviser TABLE OF CONTENTS Page INTRODUCTION Acknowledgments Statement of the Problem SECTION I REACTION OF SODIUM NITROALKANES WITH VARIOUS NITRATING AGENTS 1 I. Discussion 1 A. Introduction 1 B. Agents Investigated 2 1. Nltryl Chloride 2 a. Preparation 2 b. Review of thé Reactions of Nltryl Chloride With Organic Reagents U c. Reaction of Sodium 2-Nltropropane With Nltryl Chloride 6 d. Reaction of Sodium 2-Nltropropane with Nltryl Chloride and Aluminum Chloride 11 2. Mixed Nitric and Sulfuric Acid 12 a. Reaction of Sodium 2-Nltropropane With Mixed Nitric and Sulfuric Acid 12 3. Methyl Nitrate Ih a. Reaction of Sodium 2-Nltropropane and of Potassium Nltroethane l4 I. Alkyl Nitrates as Nitrating Agents 14 II. Discussion of Results 15 Page II. Experimental 21 A. Agents Investigated 21 1. Nltryl Chloride 21 a. Preparation 21 1. Intermediates; Chlorosulfonlc Acid and Nitric Acid, Anhydrous 11. Procedure for Making Nltryl-Chloride h. Reaction of Nltryl Chloride with a Secondary Nltroalkane 2k 1. 2-Nltropropane 11. Sodium 2-Nltropropane c. Reaction of Sodium 2-Nltropropane, Nltryl Chloride and Aluminum Chloride. 27 2. Reaction of Sodium 2-Nltropropane With Mixed Nitric and Sulfuric Acids 28 3. -

Chapter 7: Search for New Fire Suppressant Chemicals
Chapter 7: SEARCH FOR NEW FIRE J. Douglas Mather, Ph.D. SUPPRESSANT CHEMICALS Chemical Development Studies, Inc. Robert E. Tapscott, Ph.D. GlobeTech, Inc. TABLE OF CONTENTS 7.1 Fire Suppressant Replacement Knowledge Prior to the NGP .........................................612 7.1.1 Overview of Early Halon Replacement Efforts ....................................................612 7.1.2 Fire Suppressant Research – 1974 Through 1993.................................................613 7.1.3 DoD Technology Development Plan (1993 to 1997)............................................615 7.1.4 Advanced Agent Working Group (AAWG) .........................................................616 7.1.5 Summary: Alternative Agents and Selection Criteria Prior to the NGP ...............622 7.2 The NGP Approach to New Chemicals Screening..........................................................612 7.3 NGP Surveys of Inorganic Chemical Families................................................................612 7.3.1 Main Group Elements - Group I............................................................................626 7.3.2 Main Group Elements - Group II ..........................................................................627 7.3.3 Main Group Elements - Group III.........................................................................627 7.3.4 Main Group Elements - Group IV.........................................................................628 7.3.5 Main Group Elements - Group V..........................................................................634 -

What Are the Reactive Halogen Gases That Destroy Stratospheric Ozone?
What are the reactive halogen gases that destroy Q7 stratospheric ozone? The chlorine- and bromine-containing gases that enter the stratosphere arise from both human activities and natural processes. When exposed to ultraviolet radiation from the Sun, these halogen source gases are converted to more reactive gases that also contain chlorine and bromine. Some reactive gases act as chemical reservoirs which can then be converted into the most reactive gases, namely ClO and BrO. These most reactive gases participate in catalytic reactions that efficiently destroy ozone. Halogen-containing gases present in the stratosphere can be Reactive halogen gases. The chemical conversion of halogen divided into two groups: halogen source gases and reactive hal- source gases, which involves solar ultraviolet radiation and ogen gases (see Figure Q7-1). The source gases, which include other chemical reactions, produces a number of reactive halo- ozone-depleting substances (ODSs), are emitted at Earth’s gen gases. These reactive gases contain all of the chlorine and surface by natural processes and by human activities (see Q6). bromine atoms originally present in the source gases. The most Once in the stratosphere, the halogen source gases chemically important reactive chlorine- and bromine-containing gases that convert at different rates to form the reactive halogen gases. form in the stratosphere are shown in Figure Q7-1. Throughout The conversion occurs in the stratosphere instead of the tropo- the stratosphere, the most abundant are typically hydrogen sphere for most gases because solar ultraviolet radiation (a com- chloride (HCl) and chlorine nitrate (ClONO2). These two gases ponent of sunlight) is more intense in the stratosphere (see Q2). -
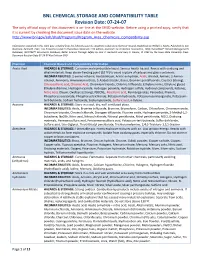
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Hydrogen Chloride-Induced Surface Disordering on Ice
Hydrogen chloride-induced surface disordering on ice V. Faye McNeill*†, Thomas Loerting‡§¶, Franz M. Geiger‡§ʈ, Bernhardt L. Trout*, and Mario J. Molina‡§** Departments of ‡Chemistry, §Earth, Atmospheric, and Planetary Sciences, and *Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 Contributed by Mario J. Molina, May 4, 2006 Characterizing the interaction of hydrogen chloride (HCl) with gas phase. We find that trace amounts of HCl induce surface polar stratospheric cloud ice particles is essential for understanding change, which we interpret to be formation of a disordered the processes responsible for ozone depletion. We studied the surface region, on ice at a range of conditions that includes interaction of gas-phase HCl with ice between 243 and 186 K by stratospherically relevant temperatures and HCl partial pres- using (i) ellipsometry to monitor the ice surface and (ii) coated-wall sures. We show that the chlorine activation reaction of HCl with flow tube experiments, both with chemical ionization mass spec- ClONO2 on ice is enhanced in the presence of surface disorder, trometry detection of the gas phase. We show that trace amounts as is the uptake of gas-phase CH3COOH. These results impact of HCl induce formation of a disordered region, or quasi-liquid our understanding of the chemistry and physics of ice particles layer, at the ice surface at stratospheric temperatures. We also in the atmosphere. show that surface disordering enhances the chlorine activation reaction of HCl with chlorine nitrate (ClONO2) and also enhances Results acetic acid (CH3COOH) adsorption. These results impact our under- Figs. 1 and 2 show the ellipsometry signals during two studies of standing of the chemistry and physics of ice particles in the ice in the presence of HCl in the gas phase, (i) at constant HCl atmosphere. -

Publicaciones
Publicaciones Molina, M.J., and G.C. Pimentel, Tandem chemical laser measurements of vibrational energy distribution in the dichloroethylene photoelimination reactions, J. Chem. Phys., 56, 3988, 1972 Molina, M.J., and G.C. Pimentel, Chemical laser studies of vibrational energy distributions: The equal-gain and zero-gain temperature techniques, IEEE J. Quantum Electronics, QE-9, 64, 1973 Molina, M.J., and F.S. Rowland, Stratospheric sink for chlorofluoromethanes-chlorine atom catalyzed destruction of ozone, Nature, 249, 810, 1974 Molina, M.J., and F.S. Rowland, Predicted present stratospheric abundances of chlorine species from photodissociation of carbon tetrachloride, Geophys. Res. Lett.,1, 309, 1974 Molina, M.J., and F.S. Rowland, Chlorofluoromethanes in the environment, Rev. Geophys. and Space Phys.,13, 1, 1975 Rowland, F.S., and M.J. Molina, Some unmeasured chlorine atom reaction rates important for stratospheric modeling of chlorine atom catalyzed removal of ozone, J. Phys. Chem., 79, 667, 1975 Rowland, F.S., and M.J. Molina, The ozone question, Science, 190, 1038, 1975 Rowland, F.S., M.J. Molina, and C.C. Chou, Natural halocarbons in air and sea, Nature, 258, 775, 1975 Rowland, F.S., and M.J. Molina, Estimated future atmospheric concentrations of CCl3F (fluorocarbon-11) for various hypothetical tropospheric removal rates, J. Phys. Chem., 80, 2049, 1976 Rowland, F.S., J.E. Spencer, and M.J. Molina, Stratospheric formation and photolysis of chlorine nitrate, ClONO2, J .Phys. Chem., 80, 2711, 1976 Rowland, F.S., J.E. Spencer, and M.J. Molina, Estimated stratospheric concentrations of chlorine nitrate, ClONO2, J .Phys. Chem., 80, 2713, 1976 Chou, C.C., W.S. -

Lemon Phd Thesis
Atmospheric Corrosion of Silver Investigated by X-ray Photoelectron Spectroscopy Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Christine Elizabeth Lemon, B.S. Graduate Program in Chemistry The Ohio State University 2012 Dissertation Committee: Heather C. Allen, Advisor Prabir K. Dutta Gerald S. Frankel Samuel D. Stout Barbara E. Wyslouzil Copyright by Christine E. Lemon 2012 Abstract Atmospheric corrosion is a costly problem. Accelerated laboratory tests, such as the salt fog chamber, have been created to predict corrosion of materials without the need to expose them over long periods of time outdoors. However, these accelerated tests often do not accurately reproduce the types and rates of corrosion found in field exposures. Silver exhibits this discrepancy and has been used in recent years in an attempt to correct the shortcomings of these accelerated tests. This study identifies Ag 2SO 3 and Ag 2SO 4 on field-exposed silver coupons. The presence of these species on field-exposed silver has been contested in the literature. Evidence suggests that Ag 2SO 3 is an intermediate step in the formation of Ag 2SO 4. Furthermore, the presence of alkali cations, such as Na +, determines the final oxidation + state of the sulfur species on silver. If Na is present, Ag 2SO 4 is the final state, whereas Ag 2SO 3 is not found in the presence of alkali cations. The identification of sulfite and/or sulfate on field-exposed samples suggests the need for further improvement of salt fog tests which do not currently include a sulfur source. -

Supplement to Comprehensive Isoprene and Terpene Chemistry Improves Simulated Surface Ozone in the Southeastern U.S
Supplement to Comprehensive isoprene and terpene chemistry improves simulated surface ozone in the southeastern U.S. Rebecca H. Schwantes1, Louisa K. Emmons1, John J. Orlando1, Mary C. Barth1, Geoffrey S. Tyndall1, Samuel R. Hall1, Kirk Ullmann1, Jason M. St. Clair2,3, Donald R. Blake4, Armin Wisthaler5,6, and ThaoPaul V. Bui7 1Atmospheric Chemistry Observations and Modeling Laboratory, National Center for Atmospheric Research, Boulder, CO 80301, U.S.A. 2Atmospheric Chemistry and Dynamics Laboratory, NASA Goddard Space Flight Center, Greenbelt, MD, 20771, USA 3Joint Center for Earth Systems Technology, University of Maryland Baltimore County, Baltimore, MD, 21228, USA 4Department of Chemistry, University of California-Irvine, 570 Rowland Hall, Irvine, CA 92697-2025, USA 5Institute for Ion Physics and Applied Physics, University of Innsbruck, Technikerstrasse 25, 6020 Innsbruck, Austria 6Department of Chemistry, University of Oslo, P.O. 1033 - Blindern, 0315 Oslo, Norway 7Earth Science Division, NASA Ames Research Center, Moffett Field, CA 94035-1000 Correspondence to: Rebecca H. Schwantes ([email protected]) Contents S1 Schematics of NO3 oxidation 3 S2 Evaluation Against More Explicit Chemical Schemes for Myrcene and β-caryophyllene 5 S3 Nudging and Vertical Level Resolution in CESM/CAM-Chem 6 5 S4 Organic Nitrate Fate in MOZART-TS1 9 S5 Tables Defining Chemical Compounds and Reactions in MOZART-TS2 10 List of Figures S1 Schematic of MOZART-TS2 isoprene NO3-initiated oxidation. .3 S2 Schematic of MOZART-TS2 terpene NO3-initiated oxidation. .4 10 S3 BOXMOX results for myrcene oxidation evaluation against explicit schemes . .5 S4 BOXMOX results for β-Caryophyllene oxidation evaluation against explicit schemes . .6 S5 SEAC4Rs flight tracks nudging tests 4km . -

Compilation of Henry's Law Constants
Compilation of Henry’s Law Constants for Inorganic and Organic Species of Potential Importance in Environmental Chemistry http://www.mpch-mainz.mpg.de/~sander/res/henry.html Rolf Sander Air Chemistry Department Max-Planck Institute of Chemistry PO Box 3060 55020 Mainz Germany e-mail: [email protected] Version 3 (April 8, 1999) c Rolf Sander (non-commercial reproduction permitted) Contents 1 Introduction 3 2 The physical quantity of solubility 3 3 Temperature dependence 3 4 Unit conversions 3 5 How to use the Tables 4 6 Further Sources of Information 4 7 Data Table (Inorganic) 6 oxygen (O) . 6 hydrogen (H) . 6 nitrogen (N) . 7 fluorine (F) . 8 chlorine (Cl) . 9 bromine (Br) . 10 iodine (I) . 11 sulfur (S) . 12 rare gases . 13 other elements . 13 2 R. Sander: Henry’s law constants (http://www.mpch-mainz.mpg.de/~sander/res/henry.html) 8 Data Table (Organic) 14 alkanes (C and H only) . 14 cycloalkanes (C and H only) . 27 aliphatic alkenes and cycloalkenes (C and H only) . 28 aliphatic alkynes (C and H only) . 30 mononuclear aromatics (C and H only) . 31 terpenes and polynuclear aromatics (C and H only) . 35 alcohols (ROH) (C, H, and O only) . 37 polyols (R(OH)n) (C, H, and O only) . 41 peroxides (ROOH) and peroxy radicals (ROO) (C, H, and O only) . 43 aldehydes (RCHO) (C, H, and O only) . 44 ketones (RCOR) (C, H, and O only) . 46 carboxylic acids (RCOOH) and peroxy carboxylic acids (RCOOOH) (C, H, and O only) . 48 esters (RCOOR) (C, H, and O only) .