Synaptic Specificity and Plasticity in Parvalbumin-Basket Cell Circuits
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cortical Bitufted, Horizontal, and Martinotti Cells Preferentially
Proc. Natl. Acad. Sci. USA Vol. 96, pp. 3217–3222, March 1999 Neurobiology Cortical bitufted, horizontal, and Martinotti cells preferentially express and secrete reelin into perineuronal nets, nonsynaptically modulating gene expression (GABAergic interneuronsyCa21-binding proteinsyneuropeptidesychandelier cellsybasket cells) CHRISTINE PESOLD*†,WEN SHENG LIU*, ALESSANDRO GUIDOTTI*, ERMINIO COSTA*, AND HECTOR J. CARUNCHO*‡ *Psychiatric Institute, Department of Psychiatry, College of Medicine, University of Illinois at Chicago, 1601 West Taylor Street, MyC 912, Chicago IL, 60612; and ‡Department of Fundamental Biology, University of Santiago de Compostela, Galicia, Spain 15706 Contributed by Erminio Costa, December 28, 1998 ABSTRACT Reelin (Reln) is a protein with some struc- mouse, shows a normal Reln expression in Cajal–Retzius cells tural analogies with other extracellular matrix proteins that and secretion in the extracellular matrix in embryonic devel- functions in the regulation of neuronal migration during the opment but neuroanatomical abnormalities reminiscent of development of cortical laminated structures. In the cortex of those in the reeler mouse (4, 5). It is currently believed that the adult animals, Reln is expressed primarily in g-aminobutyric protein encoded by this gene acts downstream of Reln in a acid (GABA)ergic neurons and is secreted into perineuronal signaling pathway that controls laminar corticogenesis and the nets. However, only 50–60% of GABAergic interneurons ex- hippocampal and cerebellar development in mammalian brain press Reln. We have characterized this subpopulation of (6). cortical GABAergic neurons that expresses Reln by using two In adult telencephalon, Reln is preferentially expressed in strategies: (i) a double immunolabeling procedure to deter- GABAergic neurons (7, 8), and Dab1 is expressed predomi- mine the colocalization of Reln with neuropeptides and Ca21- nantly in pyramidal cells, as well as in a small population of binding proteins and (ii) a combination of Golgi staining and non-pyramidal cells (9). -

A Model of Long-Term Memory Storage in the Cerebellar Cortex: a Possible Role for Plasticity at Parallel Fiber Synapses Onto Stellate͞basket Interneurons
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 14200–14205, December 1997 Psychology A model of long-term memory storage in the cerebellar cortex: A possible role for plasticity at parallel fiber synapses onto stellateybasket interneurons GARRETT T. KENYON† Department of Neurobiology and Anatomy, University of Texas Medical School at Houston, 6431 Fannin, Houston, TX 77030 Edited by Richard F. Thompson, University of Southern California, Los Angeles, CA, and approved October 3, 1997 (received for review April 14, 1997) ABSTRACT By evoking changes in climbing fiber activity, Despite evidence supporting their postulated role in motor movement errors are thought to modify synapses from parallel learning, theoretical arguments suggest that memories stored fibers onto Purkinje cells (pf*Pkj) so as to improve subse- at pf*Pkj synapses would be susceptible to long-term degra- quent motor performance. Theoretical arguments suggest dation as a result of ongoing motor adaptation. First, motor there is an intrinsic tradeoff, however, between motor adap- memories are likely to be distributed across overlapping sets of tation and long-term storage. Assuming a baseline rate of pf*Pkj synapses. If motor memories did not overlap, the motor errors is always present, then repeated performance of storage capacity of pf*Pkj synapses would be greatly reduced any learned movement will generate a series of climbing (12, 17). Second, it is reasonable to assume that the pattern of fiber-mediated corrections. By reshuffling the synaptic synaptic weights necessary to effect any given movement is weights responsible for any given movement, such corrections non-unique. If the activity of a Purkinje cell depends only on will degrade the memories for other learned movements stored the total sum of the synaptic input at any given moment, then in overlapping sets of synapses. -

Complete Dissertation
VU Research Portal Cholinergic modulation of microcircuits in the cortex Obermayer, J.M.G. 2019 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Obermayer, J. M. G. (2019). Cholinergic modulation of microcircuits in the cortex. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 04. Oct. 2021 Cholinergic modulation of microcircuits in the cortex Joshua Obermayer The research described in this thesis was conducted at the department of Integrative Neurophysiology of the Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, Vrije Universiteit Amsterdam, the Netherlands. No part of this thesis may be reproduced without prior permission of the author. Front cover: shows two pyramidal neurons and an interneuron that together form a disynaptic inhibitory microcircuit in the human cortex. -

FIRST PROOF Cerebellum
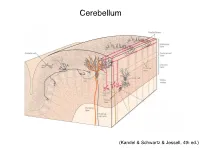
Article Number : EONS : 0736 GROSS ANATOMY Cerebellum Cortex The cerebellar cortex is an extensive three-layered sheet with a surface approximately 15 cm laterally THE HUMAN CEREBELLUM (‘‘little brain’’) is a and 180 cm rostrocaudally but densely folded around significant part of the central nervous system both three pairs of nuclei. The cortex is divided into three in size and in neural structure. It occupies approxi- transverse lobes: Anterior and posterior lobes are mately one-tenth of the cranial cavity, sitting astride separated by the primary fissure, and the smaller the brainstem, beneath the occipital cortex, and flocculonodular lobe is separated by the poster- contains more neurons than the whole of the cerebral olateral fissure (Fig. 1). The anterior and posterior cortex. It consists of an extensive cortical sheet, lobes are folded into a number of lobules and further densely folded around three pairs of nuclei. The folded into a series of folia. This transverse organiza- cortex contains only five main neural cell types and is tion is then divided at right angles into broad one of the most regular and uniform structures in the longitudinal regions. The central vermis, named for central nervous system (CNS), with an orthogonal its worm-like appearance, is most obvious in the ‘‘crystalline’’ organization. Major connections are posterior lobe. On either side is the paravermal or made to and from the spinal cord, brainstem, and intermediate cortex, which merges into the lateral sensorimotor areas of the cerebral cortex. hemispheres. The most common causes of damage to the cerebellum are stroke, tumors, or multiple sclerosis. -

And Parvalbumin-Expressing Cortical Interneurons in a Mouse Model Of
Impaired excitability of somatostatin- and PNAS PLUS parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome Chao Taia, Yasuyuki Abea,b, Ruth E. Westenbroeka, Todd Scheuera, and William A. Catteralla,1 aDepartment of Pharmacology, University of Washington, Seattle, WA 98195; and bMedicinal Safety Research Laboratories, Daiichi Sankyo Co., Ltd., Tokyo 134-8630, Japan Contributed by William A. Catterall, June 17, 2014 (sent for review May 6, 2014) Haploinsufficiency of the voltage-gated sodium channel NaV1.1 18, 19). In layer V of the cerebral cortex, PV-expressing fast- causes Dravet syndrome, an intractable developmental epilepsy spiking interneurons and SST-expressing Martinotti cells each syndrome with seizure onset in the first year of life. Specific het- account for ∼40% of interneurons, and these interneurons are erozygous deletion of NaV1.1 in forebrain GABAergic-inhibitory the major inhibitory regulators of cortical network activity (17, neurons is sufficient to cause all the manifestations of Dravet syn- 20). Layer V PV interneurons make synapses on the soma and drome in mice, but the physiological roles of specific subtypes of proximal dendrites of pyramidal neurons (18, 19), where they GABAergic interneurons in the cerebral cortex in this disease are mediate fast and powerful inhibition (21, 22). Selective hetero- unknown. Voltage-clamp studies of dissociated interneurons from zygous deletion of Scn1a in neocortical PV interneurons increases cerebral cortex did not detect a significant effect of the Dravet susceptibility to chemically induced seizures (23), spontaneous syndrome mutation on sodium currents in cell bodies. However, seizures, and premature death (24), indicating that this cell type current-clamp recordings of intact interneurons in layer V of neo- may contribute to Scn1a deficits. -

The Stellate Cells of the Rat's Cerebellar Cortex*
Z. Anat. Entwickl.-Gesch. 136, 224--248 (1972) by Springer-Verlag 1972 The Stellate Cells of the Rat's Cerebellar Cortex* Victoria Chan-Palay and Sanford L. Palay Departments of Neurobiology and Anatomy, Harvard Medical School, Boston, Massachusetts Received February 18, 1972 Summary. Stellate cells were studied in rapid Golgi preparations and in electron micro- graphs. These small neurons can be classified on the basis of their position in the molecular layer and the patterns of their dendritic and axonal arborizations as follows: (1) superficial cells with short, contorted dendrites and a circumscribed axonal arbor (upper third of the molecular layer) ; (2) deep stellate cells with radiating, twisted dendrites and with long axons giving rise to thin, varicose collaterals (middle third of the molecular layer); (3) deep stellate cells with similar dendrites and long axons giving collaterals to the basket around the Purkinje cell bodies (middle third of the molecular layer). An important characteristic of the stellate cell axon is that it generates most of its collaterals close to its origin. Even in long axon cells, only a few collaterals issue from the more distant parts of the axon. These forms contrast with the basket cell, which sends out long, straighter dendrites, and an extended axon that first emits branches at some distance from its origin. Furthermore, basket cell axon collaterals are usually stout in contrast to the frail, beaded collaterals of the stellate cell axon. The two cell types are considered to be distinct. In electron micrographs stellate cells display folded nuclei and sparse cytoplasm with the characteristics usual for small neurons. -

Or Vasoactive Intestinal Polypeptide-Containing Cells Among Gabaergic Cell Subtypes in Rat Frontal Cortex
The Journal of Neuroscience, April 15, 1996, 76(8):2701-2715 Physiological and Morphological Identification of Somatostatin- or Vasoactive Intestinal Polypeptide-Containing Cells among GABAergic Cell Subtypes in Rat Frontal Cortex Yasuo Kawaguchi and Yoshiyuki Kubota Labora tory for Neural Circuits, Bio- Mimetic Control Research Center, The Institute of Physical and Chemical Research (R/KEN), Nagoya 456, Japan Physiological and morphological characteristics of GABAer- in deep layers). Some of the Martinotti cells in layer V also gic nonpyramidal cells in frontal cortex of young rats identi- contained calbindin Dpak. VIP cells included neurons the fied immunohistochemically as containing somatostatin or main descending axons of which had more descending than vasoactive intestinal polypeptide (VIP) were studied in vitro ascending collaterals (bipolar cells and double bouquet by whole-cell recording and biocytin injection. We have cells). Two other morphological forms of the VIP cells were found that most somatostatin- or VIP-containing neurons those with short descending axons with collaterals bearing were different from two other types of GABAergic cells, the multiple boutons on other cell bodies (small basket cells) or parvalbumin-containing fast-spiking cells and the late- with short ascending main axons with collaterals forming spiking cells (neurogliaform cells). In response to injected arcades (arcade cells). Some of these neurons also con- currents, somatostatin- or VIP-containing nonpyramidal cells tained calretinin. From these results, it appears that the showed either bursts of a few spikes on a slow-depolarizing GABAergic neurons controlling circuits in the neocortical hump, burst-spiking nonpyramidal cells, or single spikes only layers may be characterized further based on whether they on depolarization, regular-spiking nonpyramidal cells. -

Layer VI in Cat Primary Auditory Cortex: Golgi Study and Sublaminar Origins of Projection Neurons
THE JOURNAL OF COMPARATIVE NEUROLOGY 404:332–358 (1999) Layer VI in Cat Primary Auditory Cortex: Golgi Study and Sublaminar Origins of Projection Neurons JORGE J. PRIETO1* AND JEFFERY A. WINER2 1Department of Histology, Institute of Neurosciences, University Miguel Herna´ ndez, 03550 San Juan, Alicante, Spain 2Division of Neurobiology, Department of Molecular and Cell Biology, University of California at Berkeley, Berkeley, California 94720–3200 ABSTRACT The organization of layer VI in cat primary auditory cortex (AI) was studied in mature specimens. Golgi-impregnated neurons were classified on the basis of their dendritic and somatic form. Ipsilateral and contralateral projection neurons and the corticogeniculate cells of origin were labeled with retrograde tracers and their profiles were compared with the results from Golgi studies. Layer VI was divided into a superficial half (layer VIa) with many pyramidal neurons and a deeper part (layer VIb) that is dominated by horizontal cells. Nine types of neuron were identified; four classes had subvarieties. Classical pyramidal cells and star, fusiform, tangential, and inverted pyramidal cells occur. Nonpyramidal neurons were Martinotti, multipolar stellate, bipolar, and horizontal cells. This variety of neurons distin- guished layer VI from other AI layers. Pyramidal neuron dendrites contributed to the vertical, modular organization in AI, although their apical processes did not project beyond layer IV. Their axons had vertical, intrinsic processes as well as corticofugal branches. Horizontal cell dendrites extended laterally up to 700 µm and could integrate thalamic input across wide expanses of the tonotopic domain. Connectional experiments confirmed the sublaminar arrangement seen in Nissl material. Commissural cells were concentrated in layer VIa, whereas corticocortical neurons were more numerous in layer VIb. -

Lateral Inhibition by Martinotti Interneurons Is Facilitated by Cholinergic Inputs in Human and Mouse Neocortex
ARTICLE DOI: 10.1038/s41467-018-06628-w OPEN Lateral inhibition by Martinotti interneurons is facilitated by cholinergic inputs in human and mouse neocortex Joshua Obermayer1, Tim S. Heistek1, Amber Kerkhofs1, Natalia A. Goriounova 1, Tim Kroon1,3, Johannes C. Baayen2, Sander Idema2, Guilherme Testa-Silva1,4, Jonathan J. Couey1,5 & Huibert D. Mansvelder 1 1234567890():,; A variety of inhibitory pathways encompassing different interneuron types shape activity of neocortical pyramidal neurons. While basket cells (BCs) mediate fast lateral inhibition between pyramidal neurons, Somatostatin-positive Martinotti cells (MCs) mediate a delayed form of lateral inhibition. Neocortical circuits are under control of acetylcholine, which is crucial for cortical function and cognition. Acetylcholine modulates MC firing, however, precisely how cholinergic inputs affect cortical lateral inhibition is not known. Here, we find that cholinergic inputs selectively augment and speed up lateral inhibition between pyramidal neurons mediated by MCs, but not by BCs. Optogenetically activated cholinergic inputs depolarize MCs through activation of ß2 subunit-containing nicotinic AChRs, not muscarinic AChRs, without affecting glutamatergic inputs to MCs. We find that these mechanisms are conserved in human neocortex. Cholinergic inputs thus enable cortical pyramidal neurons to recruit more MCs, and can thereby dynamically highlight specific circuit motifs, favoring MC- mediated pathways over BC-mediated pathways. 1 Department of Integrative Neurophysiology, Center for Neurogenomics and Cognitive Research, Neuroscience Campus, VU University Amsterdam, De Boelelaan 1085, Amsterdam 1081 HV, The Netherlands. 2 Department of Neurosurgery, Neuroscience Campus Amsterdam, VU University Medical Center Amsterdam, De Boelelaan 1117, Amsterdam 1081 HV, The Netherlands. 3Present address: MRC Centre-Developmental Neurobiology, King’s college London, London WC2R 2LS, UK. -

The Developmental Genetics of the Cerebellum and the Genetic Bases of Known Cerebellar Disorders
THE DEVELOPMENTAL GENETICS OF THE CEREBELLUM AND THE GENETIC BASES OF KNOWN CEREBELLAR DISORDERS A Review of Developmental Genetics of the Cerebellum and the Genetic Bases of known Cerebellar Disorders Olakada Favour Adamba 17/MHS01/254 Department of Medicine and Health Sciences College of Medicine and Health Sciences Afe Babalola University ANA 303: Neuroanatomy July, 2020 The Developmental Genetics of Cerebellum and the Genetic Bases of known Cerebellar Disorders: A Literature Review Olakada Favour Adamba1 Abstract The internal structure of the cerebellum is an intriguing paradox; its cytoarchitecture is relatively simple compared to the connections between its neurons, which are wired into a complex array of gene expression domains and functional circuits. The genetic research of cerebellar development has provided a great deal of information about the molecular events directing the formation of the cerebellum. The developmental mechanisms that coordinate the establishment of cerebellar structure and circuitry provide a powerful model for understanding how a functional brain develops and its significance in cerebellar disorders and diseases. The cellular makeup of the cerebellum is derived from two primary germinal matrices (the ventricular zone and a specialized germinal matrix called the rhombic lip). Each matrix/zone expresses a specific set of genes that establish the cell lineages within the cerebellar anlage. Then, cohorts of differentiated projection neurons and interneuron progenitors migrate into the developing cerebellum. thereafter, a number of remarkable patterning events occur. Altogether, structural and molecular organisations are thought to support the proper connectivity between incoming afferent projections and their target cells. Key words: Cerebellum, circuitry, genetic, development, disorders. I. Introduction The cerebellum (‘little brain’) resides at the anterior end of the hindbrain and is classically defined by its role in sensory-motor processing (Buckner, 2013). -

Revisiting Enigmatic Cortical Calretinin-Expressing Interneurons
REVIEW ARTICLE published: 24 June 2014 NEUROANATOMY doi: 10.3389/fnana.2014.00052 Revisiting enigmatic cortical calretinin-expressing interneurons Bruno Cauli 1,2,3*, Xiaojuan Zhou 4, Ludovic Tricoire 1,2,3, Xavier Toussay 1,2,3 and Jochen F. Staiger 4* 1 Sorbonne Universités, UPMC University Paris 06, UM CR18, Neuroscience Paris Seine, Paris, France 2 Centre National de la Recherche Scientifique, UMR 8246, Neuroscience Paris Seine, Paris, France 3 Institut National de la Santé et de la Recherche Médicale, UMR-S 1130, Neuroscience Paris Seine, Paris, France 4 Institute for Neuroanatomy, UMG, Georg-August-University Göttingen, Göttingen, Germany Edited by: Cortical calretinin (CR)-expressing interneurons represent a heterogeneous subpopulation Zsofia Magloczky, Hungarian of about 10–30% of GABAergic interneurons, which altogether total ca. 12–20% of all Academy of Sciences, Hungary cortical neurons. In the rodent neocortex, CR cells display different somatodendritic Reviewed by: morphologies ranging from bipolar to multipolar but the bipolar cells and their variations José A. Armengol, University Pablo de Olavide, Spain dominate. They are also diverse at the molecular level as they were shown to Fiorenzo Conti, Universita express numerous neuropeptides in different combinations including vasoactive intestinal Politecnica delle Marche, Italy polypeptide (VIP), cholecystokinin (CCK), neurokinin B (NKB) corticotrophin releasing *Correspondence: factor (CRF), enkephalin (Enk) but also neuropeptide Y (NPY) and somatostatin (SOM) Bruno Cauli, CNRS-UPMC, to a lesser extent. CR-expressing interneurons exhibit different firing behaviors such as Neuroscience Paris Seine, 9 Quai Saint Bernard, 75005 Paris, France adapting, bursting or irregular. They mainly originate from the caudal ganglionic eminence e-mail: [email protected]; (CGE) but a subpopulation also derives from the dorsal part of the medial ganglionic Jochen F.Staiger, Institute for eminence (MGE). -
Cerebellum and Activation of the Cerebellum IO ST During Nonmotor Tasks
Cerebellum (Kandel & Schwartz & Jessell, 4th ed.) Granule186 cells are irreducibly smallChapter (6-8 7 μm) stellate cell basket cell outer synaptic layer PC rs cap PC layer grc Go inner 6 μm synaptic layer Figure 7.15 Largest cerebellar neuron occupies more than a 1,000-fold greater volume than smallest neuron. Thin section (~1 μ m) through monkey cerebellar cortex. Purkinje cell body (PC) and nucleus are far larger than those of granule cell (grc). The latter cluster to leave space for mossy fiber terminals to form glomeruli with grc dendritic claws and space for Golgi cells (Go). Note rich network of capillaries (cap). Fine, scat- tered dots are mitochondria. Courtesy of E. Mugnaini. brain ’ s most numerous neuron, this small cost grows large (see also chapter 13). Much of the inner synaptic layer is occupied by the large axon terminals of mossy fibers (figures 7.1 and 7.16). A terminal interlaces with multiple (~15) dendritic claws, each from a different but neighboring granule cell, and forms a complex knot (glomerulus ), nearly as large as a granule cell body (figures 7.1 and 7.16). The mossy fiber axon fires at an unusually high mean rate (up to 200 Hz) and is therefore among the brain’ s thickest (figure 4.6). To match the axon’ s high rate, a terminal expresses 150 active zones, 10 per postsynaptic granule cell (figure 7.16). These sites are capable of driving … and most expensive. 190 Chapter 7 10 4 8 3 9 19 10 10 × 6 × 2 2 4 ATP/s/cell ATP/s/cell ATP/s/m 1 2 0 0 astrocyte astrocyte Golgi cell Golgi cell basket cell basket cell stellate cell stellate cell granule cell granule cell mossy fiber mossy fiber Purkinje cell Purkinje Purkinje cell Purkinje Bergman glia Bergman glia climbing fiber climbing fiber Figure 7.18 Energy costs by cell type.