Colletotrichum Graminicola</Em>
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptomic Characterization of Bradyrhizobium Diazoefficiens
International Journal of Molecular Sciences Article Transcriptomic Characterization of Bradyrhizobium diazoefficiens Bacteroids Reveals a Post-Symbiotic, Hemibiotrophic-Like Lifestyle of the Bacteria within Senescing Soybean Nodules Sooyoung Franck 1, Kent N. Strodtman 1 , Jing Qiu 2 and David W. Emerich 1,* 1 Division of Biochemistry, University of Missouri, Columbia, MO 65211, USA; [email protected] (S.F.); [email protected] (K.N.S.) 2 Applied Economics and Statistics, University of Delaware, Newark, DE 19716, USA; [email protected] * Correspondence: [email protected]; Tel: +1-573-882-4252 Received: 8 October 2018; Accepted: 28 November 2018; Published: 7 December 2018 Abstract: The transcriptional activity of Bradyrhizobium diazoefficens isolated from soybean nodules was monitored over the period from symbiosis to late plant nodule senescence. The bacteria retained a near constant level of RNA throughout this period, and the variation in genes demonstrating increased, decreased, and/or patterned transcriptional activity indicates that the bacteria are responding to the changing environment within the nodule as the plant cells progress from an organized cellular structure to an unorganized state of internal decay. The transcriptional variation and persistence of the bacteria suggest that the bacteria are adapting to their environment and acting similar to hemibiotrophs, which survive both as saprophytes on live plant tissues and then as necrophytes on decaying plant tissues. The host plant restrictions of symbiosis make B. diazoefficiens a highly specialized, restricted hemibiotroph. Keywords: bradyrhizobium diazoefficiens; soybean; Glycine max; nitrogen fixation; senescence; transcriptomics; hemibiotroph 1. Introduction Soybean nodules are symbiotic organs that are formed on roots by the complex interaction between soybean plants and rhizobia, nitrogen-fixing bacteria, under nitrogen-limiting conditions. -

University of London Thesis
REFERENCE ONLY UNIVERSITY OF LONDON THESIS Degree Year^^0^ Name of Author C O P Y R IG H T This is a thesis accepted for a Higher Degree of the University of London. It is an unpublished typescript and the copyright is held by the author. All persons consulting the thesis must read and abide by the Copyright Declaration below. COPYRIGHT DECLARATION I recognise that the copyright of the above-described thesis rests with the author and that no quotation from it or information derived from it may be published without the prior written consent of the author. LOANS Theses may not be lent to individuals, but the Senate House Library may lend a copy to approved libraries within the United Kingdom, for consultation solely on the premises of those libraries. Application should be made to: Inter-Library Loans, Senate House Library, Senate House, Malet Street, London WC1E 7HU. REPRODUCTION University of London theses may not be reproduced without explicit written permission from the Senate House Library. Enquiries should be addressed to the Theses Section of the Library. Regulations concerning reproduction vary according to the date of acceptance of the thesis and are listed below as guidelines. A. Before 1962. Permission granted only upon the prior written consent of the author. (The Senate House Library will provide addresses where possible). B. 1962- 1974. In many cases the author has agreed to permit copying upon completion of a Copyright Declaration. C. 1975 - 1988. Most theses may be copied upon completion of a Copyright Declaration. D. 1989 onwards. Most theses may be copied. -

257 Absolute Configuration 39, 75 – of L-(+)-Alanine 83
257 Index a L-aminoacylase 246 absolute configuration 39, 75 amount of substance 12 –ofL-(+)-alanine 83 asymmetric atom 39 – by anomalous dispersion effect in X-ray asymmetric disequilibrating transformation crystallography 78 151 – by chemical correlation 79 asymmetric induction 143 – correlation strategies 80 – of second kind 151 – by direct methods 78 asymmetric synthesis 139 – extended sense of 77 asymmetric transformation –ofD-glyceraldehyde 39 – of second kind 151 –ofD-(+)-glyceraldehyde 77 asymmetric transformation of the second kind – by indirect methods 79 173 –ofD-(–)-lactic acid 81 atropisomer 44, 73 – methods of determination 78 atropisomerism 24, 71 – of natural glucose 78, 82 autocatalysis – by predictive calculation of chiroptical data – a special case of organocatalysis 234 79 autocatalytic effect –of(R,R)-(+)-tartaric acid 78, 82 – of a zinc complex 238 achiral bidentate reagent Avogadro constant 12 – purification of enantiomers with 149 axial chirality 39, 44 achiral stationary phase 95 achirality, achiral 24, 29 b aci form 113 Baeyer–Villiger oxidation acylase I 161 – enanatiotope selective 253 AD-mix- 229 – enzymatic 253 AD-mix- 229 – microbial 166 alcohol dehydrogenases – regio- and enanatiomer selective 253 – Prelog’s rule 252 baker’s yeast aldol reactions – regio-, enantiotope and diastereotope – catalyzed with chiral ionic liquid 238 selective reduction with 186 – catalytic, double enantiotope selective 238 bidentate achiral reagent 90 – completely syn diastereoselective 238 Bijvoet, J. M. 78 – stereoselctive 209 BINAL-H 212 allene 44 BINAP amide-imido acid tautomerism 112 – ruthenium complex of 214 amino acids biocatalysis 244 enantiopure, D-andL- 250 – advantages of 245 D-amino acid oxidase 100 – disadvantages of 245 Stereochemistry and Stereoselective Synthesis: An Introduction, First Edition. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Biocatalytic application of rare PLP- dependent aminotransferases for the synthesis of high value amino acids and amines Annabel Serpico A Thesis Submitted for the Degree of Doctor of Philosophy The University of Edinburgh 2018 Lay Summary Traditional chemical approaches for the production of compounds often rely on harsh conditions such as high temperature and pressure and create toxic waste and pollutants. Moreover, these reactions are often non efficient, creating unwanted side products that need to be disposed of. A very important branch of chemistry, called green chemistry, supports the design of products and processes that use the least amount of dangerous substances as they can. An important aspect of green chemistry is biocatalysis, which consists of the use of biological systems, either whole cells or enzymes for the production of compounds. -

Of Pseudomonas Sp. Strain NS671 Expressed In
JOURNAL OF BACTERIOLOGY, Dec. 1992, p. 7989-7995 Vol. 174, No. 24 0021-9193/92/247989-07$02.00/0 Copyright X) 1992, American Society for Microbiology Purification and Characterization of the Hydantoin Racemase of Pseudomonas sp. Strain NS671 Expressed in Escherichia coli KEN WATABE,* TAKAHIRO ISHIKAWA, YUKUO MUKOHARA, AND HIROAKI NAKAMURA Odawara Research Center, Nippon Soda Co., Ltd., 345 Takada, Odawara, Kanagawa 250-02, Japan Received 16 July 1992/Accepted 19 October 1992 The hydantoin racemase gene ofPseudomonas sp. strain NS671 had been cloned and expressed in Escherichia coli. Hydantoin racemase was purified from the cell extract of the E. coli strain by phenyl-Sepharose, DEAE-Sephacel, and Sephadex G-200 chromatographies. The purified enzyme had an apparent molecular mass of 32 kDa as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. By gel filtration, a molecular mass of about 190 kDa was found, suggesting that the native enzyme is a hexamer. The optimal conditions for hydantoin racemase activity were pH 9.5 and a temperature of 45°C. The enzyme activity was slightly stimulated by the addition of not only Mn2+ or Co2' but also metal-chelating agents, indicating that the enzyme is not a metalloenzyme. On the other hand, Cu2' and Zn2+ strongly inhibited the enzyme activity. Kinetic studies showed substrate inhibition, and the V.. values for D- and L-5-(2-methylthioethyl)hydantoin were 35.2 and 79.0 ,umol/min/mg of protein, respectively. The purified enzyme did not racemize 5-isopropyl- hydantoin, whereas the cells ofE. coli expressing the enzyme are capable of racemizing it. -
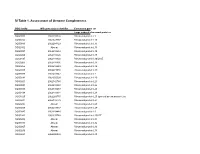
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Assay of the Prostate Cancer Biomarker A-Methylacyl Coenzyme a Racemase (AMACR)
Assay of the Prostate Cancer Biomarker a-Methylacyl Coenzyme A Racemase (AMACR) by Dahmane Ouazia Submitted in partial fulfillment of the requirements for the degree of Master of Science at Dalhousie University Halifax, Nova Scotia June 2008 © Copyright by Dahmane Ouazia, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-43994-4 Our file Notre reference ISBN: 978-0-494-43994-4 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -

The Chemistry and Evolution of Enzyme Function
The Chemistry and Evolution of Enzyme Function: Isomerases as a Case Study Sergio Mart´ınez Cuesta EMBL - European Bioinformatics Institute Gonville and Caius College University of Cambridge A thesis submitted for the degree of Doctor of Philosophy 31st July 2014 This dissertation is the result of my own work and contains nothing which is the outcome of work done in collaboration except where specifically indicated in the text. No part of this dissertation has been submitted or is currently being submitted for any other degree or diploma or other qualification. This thesis does not exceed the specified length limit of 60.000 words as defined by the Biology Degree Committee. This thesis has been typeset in 12pt font using LATEX according to the specifications de- fined by the Board of Graduate Studies and the Biology Degree Committee. Cambridge, 31st July 2014 Sergio Mart´ınezCuesta To my parents and my sister Contents Abstract ix Acknowledgements xi List of Figures xiii List of Tables xv List of Publications xvi 1 Introduction 1 1.1 Chemistry of enzymes . .2 1.1.1 Catalytic sites, mechanisms and cofactors . .3 1.1.2 Enzyme classification . .5 1.2 Evolution of enzyme function . .6 1.3 Similarity between enzymes . .8 1.3.1 Comparing sequences and structures . .8 1.3.2 Comparing genomic context . .9 1.3.3 Comparing biochemical reactions and mechanisms . 10 1.4 Isomerases . 12 1.4.1 Metabolism . 13 1.4.2 Genome . 14 1.4.3 EC classification . 15 1.4.4 Applications . 18 1.5 Structure of the thesis . 20 2 Data Resources and Methods 21 2.1 Introduction . -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

IDENTIFICATION of NATIVE 20S PROTEASOMAL SUBSTRATES in the HALOARCHAEON Haloferax Volcanii THROUGH DEGRADOMIC and PHOSPHOPROTEOMIC ANALYSIS
IDENTIFICATION OF NATIVE 20S PROTEASOMAL SUBSTRATES IN THE HALOARCHAEON Haloferax volcanii THROUGH DEGRADOMIC AND PHOSPHOPROTEOMIC ANALYSIS By PHILLIP A. KIRKLAND A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2007 1 © 2007 Phillip Kirkland 2 To my mother, Kelly Kirkland; my grandmother Judy Jansen; and to the loving memory of Mr. and Mrs. L.V. Stevens. 3 ACKNOWLEDGMENTS I would like to express my deepest gratitude to Dr. Julie Maupin for her patience and guidance. Many thanks go to the Maupin lab staff (Matt Humbard, Wei Li, and Guanguin Zhou) and to former graduates Dr. Chris Reuter and Gosia Gil for all of their help and support. My sincerest thanks go to my supervisory committee (Dr. James Preston, Dr. Madeline Rasche, Dr. Richard Lamont, Dr. Peter Kima and Dr. Eric Triplett) for their insight. I owe a debt of gratitude to John Moore, Phi Do, Dr. Franz St. John, Youngnyun Kim, John Rice and the entire Microbiology and Cell Science Department. I am also extremely grateful to all of those who have offered help and support with instrumentation, bioinformatics and general proteomic endeavors including Dr. Stan Stevens, Scott McClung, Dr. Marjorie Chow and Dr. Alfred Chung and the entire ICBR staff as well as Dr. Jennifer Busby and Valerie Cavett at Scripps Florida. Finally, I am eternally grateful for the support and encouragement of my family and friends: my mom, Kelly Kirkland, my grandmother Judy Jansen, my fiancé Shelley Cooper, Jeremy and Matt Jones, William Knapp, and the Smith, Bright, Blair, Stevens and Jansen families and my great grandparents, the late Lavert V. -

Row Labels Gene Name GLU 1 GLU 2 GLU 3 RF 1 RF 2 RF 3
Row Labels Gene Name GLU 1 GLU 2 GLU 3 RF 1 RF 2 RF 3 Ga0073285_102189 S-layer homology domain-containing protein 1549 1805 1683 1214 1230 1364 Ga0073285_10263 pyruvate-ferredoxin/flavodoxin oxidoreductase 901 1003 950 757 754 738 Ga0073285_14010 acetaldehyde dehydrogenase /alcohol 774 880 805 751 771 823 dehydrogenase AdhE Ga0073285_1166 chaperonin GroEL 663 798 645 563 703 716 Ga0073285_11916 enolase 486 378 459 422 454 412 Ga0073285_11684 elongation factor Tu 426 390 479 453 396 361 Ga0073285_11912 glyceraldehyde 3-phosphate dehydrogenase 443 382 352 417 381 367 Ga0073285_101203 ketol-acid reductoisomerase 465 555 441 251 251 280 Ga0073285_10199 4-hydroxy-3-methylbut-2-enyl diphosphate 361 382 343 314 358 372 reductase Ga0073285_102163 ATP synthase F1 subcomplex beta subunit 256 228 255 241 272 295 Ga0073285_1376 DNA-directed RNA polymerase subunit beta' 195 227 186 155 161 171 Ga0073285_12917 D-3-phosphoglycerate dehydrogenase 237 195 192 142 147 138 Ga0073285_10829 pyruvate carboxylase 185 226 195 141 148 151 Ga0073285_1377 DNA-directed RNA polymerase subunit beta 185 212 179 144 145 157 Ga0073285_11620 NAD(P)-dependent iron-only hydrogenase catalytic 98 91 93 233 227 248 subunit Contaminant_TRYP_BOVIN #N/A 157 200 188 130 150 149 Ga0073285_11681 LSU ribosomal protein L4P 198 217 190 108 122 131 Ga0073285_11561 carbamoyl-phosphate synthase large subunit 184 186 149 141 147 142 Ga0073285_101306 polyribonucleotide nucleotidyltransferase 165 164 147 128 146 162 Ga0073285_102105 inosine-5'-monophosphate dehydrogenase 221 178 182 95 116