Lecture Notes, Statistical Mechanics (Theory F)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lab: Specific Heat
Lab: Specific Heat Summary Students relate thermal energy to heat capacity by comparing the heat capacities of different materials and graphing the change in temperature over time for a specific material. Students explore the idea of insulators and conductors in an attempt to apply their knowledge in real world applications, such as those that engineers would. Engineering Connection Engineers must understand the heat capacity of insulators and conductors if they wish to stay competitive in “green” enterprises. Engineers who design passive solar heating systems take advantage of the natural heat capacity characteristics of materials. In areas of cooler climates engineers chose building materials with a low heat capacity such as slate roofs in an attempt to heat the home during the day. In areas of warmer climates tile roofs are popular for their ability to store the sun's heat energy for an extended time and release it slowly after the sun sets, to prevent rapid temperature fluctuations. Engineers also consider heat capacity and thermal energy when they design food containers and household appliances. Just think about your aluminum soda can versus glass bottles! Grade Level: 9-12 Group Size: 4 Time Required: 90 minutes Activity Dependency :None Expendable Cost Per Group : US$ 5 The cost is less if thermometers are available for each group. NYS Science Standards: STANDARD 1 Analysis, Inquiry, and Design STANDARD 4 Performance indicator 2.2 Keywords: conductor, energy, energy storage, heat, specific heat capacity, insulator, thermal energy, thermometer, thermocouple Learning Objectives After this activity, students should be able to: Define heat capacity. Explain how heat capacity determines a material's ability to store thermal energy. -

Thermodynamics Notes
Thermodynamics Notes Steven K. Krueger Department of Atmospheric Sciences, University of Utah August 2020 Contents 1 Introduction 1 1.1 What is thermodynamics? . .1 1.2 The atmosphere . .1 2 The Equation of State 1 2.1 State variables . .1 2.2 Charles' Law and absolute temperature . .2 2.3 Boyle's Law . .3 2.4 Equation of state of an ideal gas . .3 2.5 Mixtures of gases . .4 2.6 Ideal gas law: molecular viewpoint . .6 3 Conservation of Energy 8 3.1 Conservation of energy in mechanics . .8 3.2 Conservation of energy: A system of point masses . .8 3.3 Kinetic energy exchange in molecular collisions . .9 3.4 Working and Heating . .9 4 The Principles of Thermodynamics 11 4.1 Conservation of energy and the first law of thermodynamics . 11 4.1.1 Conservation of energy . 11 4.1.2 The first law of thermodynamics . 11 4.1.3 Work . 12 4.1.4 Energy transferred by heating . 13 4.2 Quantity of energy transferred by heating . 14 4.3 The first law of thermodynamics for an ideal gas . 15 4.4 Applications of the first law . 16 4.4.1 Isothermal process . 16 4.4.2 Isobaric process . 17 4.4.3 Isosteric process . 18 4.5 Adiabatic processes . 18 5 The Thermodynamics of Water Vapor and Moist Air 21 5.1 Thermal properties of water substance . 21 5.2 Equation of state of moist air . 21 5.3 Mixing ratio . 22 5.4 Moisture variables . 22 5.5 Changes of phase and latent heats . -

2D Potts Model Correlation Lengths
KOMA Revised version May D Potts Mo del Correlation Lengths Numerical Evidence for = at o d t Wolfhard Janke and Stefan Kappler Institut f ur Physik Johannes GutenbergUniversitat Mainz Staudinger Weg Mainz Germany Abstract We have studied spinspin correlation functions in the ordered phase of the twodimensional q state Potts mo del with q and at the rstorder transition p oint Through extensive Monte t Carlo simulations we obtain strong numerical evidence that the cor relation length in the ordered phase agrees with the exactly known and recently numerically conrmed correlation length in the disordered phase As a byproduct we nd the energy moments o t t d in the ordered phase at in very go o d agreement with a recent large t q expansion PACS numbers q Hk Cn Ha hep-lat/9509056 16 Sep 1995 Introduction Firstorder phase transitions have b een the sub ject of increasing interest in recent years They play an imp ortant role in many elds of physics as is witnessed by such diverse phenomena as ordinary melting the quark decon nement transition or various stages in the evolution of the early universe Even though there exists already a vast literature on this sub ject many prop erties of rstorder phase transitions still remain to b e investigated in detail Examples are nitesize scaling FSS the shap e of energy or magnetization distributions partition function zeros etc which are all closely interrelated An imp ortant approach to attack these problems are computer simulations Here the available system sizes are necessarily -

Full and Unbiased Solution of the Dyson-Schwinger Equation in the Functional Integro-Differential Representation
PHYSICAL REVIEW B 98, 195104 (2018) Full and unbiased solution of the Dyson-Schwinger equation in the functional integro-differential representation Tobias Pfeffer and Lode Pollet Department of Physics, Arnold Sommerfeld Center for Theoretical Physics, University of Munich, Theresienstrasse 37, 80333 Munich, Germany (Received 17 March 2018; revised manuscript received 11 October 2018; published 2 November 2018) We provide a full and unbiased solution to the Dyson-Schwinger equation illustrated for φ4 theory in 2D. It is based on an exact treatment of the functional derivative ∂/∂G of the four-point vertex function with respect to the two-point correlation function G within the framework of the homotopy analysis method (HAM) and the Monte Carlo sampling of rooted tree diagrams. The resulting series solution in deformations can be considered as an asymptotic series around G = 0 in a HAM control parameter c0G, or even a convergent one up to the phase transition point if shifts in G can be performed (such as by summing up all ladder diagrams). These considerations are equally applicable to fermionic quantum field theories and offer a fresh approach to solving functional integro-differential equations beyond any truncation scheme. DOI: 10.1103/PhysRevB.98.195104 I. INTRODUCTION was not solved and differences with the full, exact answer could be seen when the correlation length increases. Despite decades of research there continues to be a need In this work, we solve the full DSE by writing them as a for developing novel methods for strongly correlated systems. closed set of integro-differential equations. Within the HAM The standard Monte Carlo approaches [1–5] are convergent theory there exists a semianalytic way to treat the functional but suffer from a prohibitive sign problem, scaling exponen- derivatives without resorting to an infinite expansion of the tially in the system volume [6]. -

Compression of an Ideal Gas with Temperature Dependent Specific
Session 3666 Compression of an Ideal Gas with Temperature-Dependent Specific Heat Capacities Donald W. Mueller, Jr., Hosni I. Abu-Mulaweh Engineering Department Indiana University–Purdue University Fort Wayne Fort Wayne, IN 46805-1499, USA Overview The compression of gas in a steady-state, steady-flow (SSSF) compressor is an important topic that is addressed in virtually all engineering thermodynamics courses. A typical situation encountered is when the gas inlet conditions, temperature and pressure, are known and the compressor dis- charge pressure is specified. The traditional instructional approach is to make certain assumptions about the compression process and about the gas itself. For example, a special case often consid- ered is that the compression process is reversible and adiabatic, i.e. isentropic. The gas is usually ÊÌ considered to be ideal, i.e. ÈÚ applies, with either constant or temperature-dependent spe- cific heat capacities. The constant specific heat capacity assumption allows for direct computation of the discharge temperature, while the temperature-dependent specific heat assumption does not. In this paper, a MATLAB computer model of the compression process is presented. The substance being compressed is air which is considered to be an ideal gas with temperature-dependent specific heat capacities. Two approaches are presented to describe the temperature-dependent properties of air. The first approach is to use the ideal gas table data from Ref. [1] with a look-up interpolation scheme. The second approach is to use a curve fit with the NASA Lewis coefficients [2,3]. The computer model developed makes use of a bracketing–bisection algorithm [4] to determine the temperature of the air after compression. -
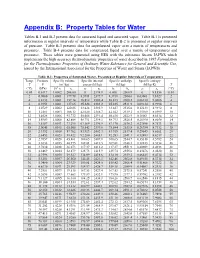
Appendix B: Property Tables for Water
Appendix B: Property Tables for Water Tables B-1 and B-2 present data for saturated liquid and saturated vapor. Table B-1 is presented information at regular intervals of temperature while Table B-2 is presented at regular intervals of pressure. Table B-3 presents data for superheated vapor over a matrix of temperatures and pressures. Table B-4 presents data for compressed liquid over a matrix of temperatures and pressures. These tables were generated using EES with the substance Steam_IAPWS which implements the high accuracy thermodynamic properties of water described in 1995 Formulation for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific Use, issued by the International Associated for the Properties of Water and Steam (IAPWS). Table B-1: Properties of Saturated Water, Presented at Regular Intervals of Temperature Temp. Pressure Specific volume Specific internal Specific enthalpy Specific entropy T P (m3/kg) energy (kJ/kg) (kJ/kg) (kJ/kg-K) T 3 (°C) (kPa) 10 vf vg uf ug hf hg sf sg (°C) 0.01 0.6117 1.0002 206.00 0 2374.9 0.000 2500.9 0 9.1556 0.01 2 0.7060 1.0001 179.78 8.3911 2377.7 8.3918 2504.6 0.03061 9.1027 2 4 0.8135 1.0001 157.14 16.812 2380.4 16.813 2508.2 0.06110 9.0506 4 6 0.9353 1.0001 137.65 25.224 2383.2 25.225 2511.9 0.09134 8.9994 6 8 1.0729 1.0002 120.85 33.626 2385.9 33.627 2515.6 0.12133 8.9492 8 10 1.2281 1.0003 106.32 42.020 2388.7 42.022 2519.2 0.15109 8.8999 10 12 1.4028 1.0006 93.732 50.408 2391.4 50.410 2522.9 0.18061 8.8514 12 14 1.5989 1.0008 82.804 58.791 2394.1 58.793 2526.5 -

A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Article A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action Ivan Kennedy 1,2,*, Harold Geering 2, Michael Rose 3 and Angus Crossan 2 1 Sydney Institute of Agriculture, University of Sydney, NSW 2006, Australia 2 QuickTest Technologies, PO Box 6285 North Ryde, NSW 2113, Australia; [email protected] (H.G.); [email protected] (A.C.) 3 NSW Department of Primary Industries, Wollongbar NSW 2447, Australia; [email protected] * Correspondence: [email protected]; Tel.: + 61-4-0794-9622 Received: 23 March 2019; Accepted: 26 April 2019; Published: 1 May 2019 Abstract: A convenient practical model for accurately estimating the total entropy (ΣSi) of atmospheric gases based on physical action is proposed. This realistic approach is fully consistent with statistical mechanics, but reinterprets its partition functions as measures of translational, rotational, and vibrational action or quantum states, to estimate the entropy. With all kinds of molecular action expressed as logarithmic functions, the total heat required for warming a chemical system from 0 K (ΣSiT) to a given temperature and pressure can be computed, yielding results identical with published experimental third law values of entropy. All thermodynamic properties of gases including entropy, enthalpy, Gibbs energy, and Helmholtz energy are directly estimated using simple algorithms based on simple molecular and physical properties, without resource to tables of standard values; both free energies are measures of quantum field states and of minimal statistical degeneracy, decreasing with temperature and declining density. We propose that this more realistic approach has heuristic value for thermodynamic computation of atmospheric profiles, based on steady state heat flows equilibrating with gravity. -

The Emergence of a Classical World in Bohmian Mechanics
The Emergence of a Classical World in Bohmian Mechanics Davide Romano Lausanne, 02 May 2014 Structure of QM • Physical state of a quantum system: - (normalized) vector in Hilbert space: state-vector - Wavefunction: the state-vector expressed in the coordinate representation Structure of QM • Physical state of a quantum system: - (normalized) vector in Hilbert space: state-vector; - Wavefunction: the state-vector expressed in the coordinate representation; • Physical properties of the system (Observables): - Observable ↔ hermitian operator acting on the state-vector or the wavefunction; - Given a certain operator A, the quantum system has a definite physical property respect to A iff it is an eigenstate of A; - The only possible measurement’s results of the physical property corresponding to the operator A are the eigenvalues of A; - Why hermitian operators? They have a real spectrum of eigenvalues → the result of a measurement can be interpreted as a physical value. Structure of QM • In the general case, the state-vector of a system is expressed as a linear combination of the eigenstates of a generic operator that acts upon it: 퐴 Ψ = 푎 Ψ1 + 푏|Ψ2 • When we perform a measure of A on the system |Ψ , the latter randomly collapses or 2 2 in the state |Ψ1 with probability |푎| or in the state |Ψ2 with probability |푏| . Structure of QM • In the general case, the state-vector of a system is expressed as a linear combination of the eigenstates of a generic operator that acts upon it: 퐴 Ψ = 푎 Ψ1 + 푏|Ψ2 • When we perform a measure of A on the system |Ψ , the latter randomly collapses or 2 2 in the state |Ψ1 with probability |푎| or in the state |Ψ2 with probability |푏| . -

Aspects of Loop Quantum Gravity
Aspects of loop quantum gravity Alexander Nagen 23 September 2020 Submitted in partial fulfilment of the requirements for the degree of Master of Science of Imperial College London 1 Contents 1 Introduction 4 2 Classical theory 12 2.1 The ADM / initial-value formulation of GR . 12 2.2 Hamiltonian GR . 14 2.3 Ashtekar variables . 18 2.4 Reality conditions . 22 3 Quantisation 23 3.1 Holonomies . 23 3.2 The connection representation . 25 3.3 The loop representation . 25 3.4 Constraints and Hilbert spaces in canonical quantisation . 27 3.4.1 The kinematical Hilbert space . 27 3.4.2 Imposing the Gauss constraint . 29 3.4.3 Imposing the diffeomorphism constraint . 29 3.4.4 Imposing the Hamiltonian constraint . 31 3.4.5 The master constraint . 32 4 Aspects of canonical loop quantum gravity 35 4.1 Properties of spin networks . 35 4.2 The area operator . 36 4.3 The volume operator . 43 2 4.4 Geometry in loop quantum gravity . 46 5 Spin foams 48 5.1 The nature and origin of spin foams . 48 5.2 Spin foam models . 49 5.3 The BF model . 50 5.4 The Barrett-Crane model . 53 5.5 The EPRL model . 57 5.6 The spin foam - GFT correspondence . 59 6 Applications to black holes 61 6.1 Black hole entropy . 61 6.2 Hawking radiation . 65 7 Current topics 69 7.1 Fractal horizons . 69 7.2 Quantum-corrected black hole . 70 7.3 A model for Hawking radiation . 73 7.4 Effective spin-foam models . -
![Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals](https://docslib.b-cdn.net/cover/4942/arxiv-1910-10745v1-cond-mat-str-el-23-oct-2019-2-2-symmetry-protected-time-crystals-304942.webp)
Arxiv:1910.10745V1 [Cond-Mat.Str-El] 23 Oct 2019 2.2 Symmetry-Protected Time Crystals
A Brief History of Time Crystals Vedika Khemania,b,∗, Roderich Moessnerc, S. L. Sondhid aDepartment of Physics, Harvard University, Cambridge, Massachusetts 02138, USA bDepartment of Physics, Stanford University, Stanford, California 94305, USA cMax-Planck-Institut f¨urPhysik komplexer Systeme, 01187 Dresden, Germany dDepartment of Physics, Princeton University, Princeton, New Jersey 08544, USA Abstract The idea of breaking time-translation symmetry has fascinated humanity at least since ancient proposals of the per- petuum mobile. Unlike the breaking of other symmetries, such as spatial translation in a crystal or spin rotation in a magnet, time translation symmetry breaking (TTSB) has been tantalisingly elusive. We review this history up to recent developments which have shown that discrete TTSB does takes place in periodically driven (Floquet) systems in the presence of many-body localization (MBL). Such Floquet time-crystals represent a new paradigm in quantum statistical mechanics — that of an intrinsically out-of-equilibrium many-body phase of matter with no equilibrium counterpart. We include a compendium of the necessary background on the statistical mechanics of phase structure in many- body systems, before specializing to a detailed discussion of the nature, and diagnostics, of TTSB. In particular, we provide precise definitions that formalize the notion of a time-crystal as a stable, macroscopic, conservative clock — explaining both the need for a many-body system in the infinite volume limit, and for a lack of net energy absorption or dissipation. Our discussion emphasizes that TTSB in a time-crystal is accompanied by the breaking of a spatial symmetry — so that time-crystals exhibit a novel form of spatiotemporal order. -

Particle Characterisation In
LIFE SCIENCE I TECHNICAL BULLETIN ISSUE N°11 /JULY 2008 PARTICLE CHARACTERISATION IN EXCIPIENTS, DRUG PRODUCTS AND DRUG SUBSTANCES AUTHOR: HILDEGARD BRÜMMER, PhD, CUSTOMER SERVICE MANAGER, SGS LIFE SCIENCE SERVICES, GERMANY Particle characterization has significance in many industries. For the pharmaceutical industry, particle size impacts products in two ways: as an influence on drug performance and as an indicator of contamination. This article will briefly examine how particle size impacts both, and review the arsenal of methods for measuring and tracking particle size. Furthermore, examples of compromised product quality observed within our laboratories illustrate why characterization is so important. INDICATOR OF CONTAMINATION Controlling the limits of contaminating • Adverse indirect reactions: particles Particle characterisation of drug particles is critical for injectable are identified by the immune system as substances, drug products and excipients (parenteral) solutions. Particle foreign material and immune reaction is an important factor in R&D, production contamination of solutions can potentially might impose secondary effects. and quality control of pharmaceuticals. have the following results: It is becoming increasingly important for In order to protect a patient and to compliance with requirements of FDA and • Adverse direct reactions: e.g. particles guarantee a high quality product, several European Health Authorities. are distributed via the blood in the body chapters in the compendia (USP, EP, JP) and cause toxicity to specific tissues or describe techniques for characterisation organs, or particles of a given size can of limits. Some of the most relevant cause a physiological effect blocking blood chapters are listed in Table 1. flow e.g. in the lungs. -

LECTURES on MATHEMATICAL STATISTICAL MECHANICS S. Adams
ISSN 0070-7414 Sgr´ıbhinn´ıInstiti´uid Ard-L´einnBhaile´ Atha´ Cliath Sraith. A. Uimh 30 Communications of the Dublin Institute for Advanced Studies Series A (Theoretical Physics), No. 30 LECTURES ON MATHEMATICAL STATISTICAL MECHANICS By S. Adams DUBLIN Institi´uid Ard-L´einnBhaile´ Atha´ Cliath Dublin Institute for Advanced Studies 2006 Contents 1 Introduction 1 2 Ergodic theory 2 2.1 Microscopic dynamics and time averages . .2 2.2 Boltzmann's heuristics and ergodic hypothesis . .8 2.3 Formal Response: Birkhoff and von Neumann ergodic theories9 2.4 Microcanonical measure . 13 3 Entropy 16 3.1 Probabilistic view on Boltzmann's entropy . 16 3.2 Shannon's entropy . 17 4 The Gibbs ensembles 20 4.1 The canonical Gibbs ensemble . 20 4.2 The Gibbs paradox . 26 4.3 The grandcanonical ensemble . 27 4.4 The "orthodicity problem" . 31 5 The Thermodynamic limit 33 5.1 Definition . 33 5.2 Thermodynamic function: Free energy . 37 5.3 Equivalence of ensembles . 42 6 Gibbs measures 44 6.1 Definition . 44 6.2 The one-dimensional Ising model . 47 6.3 Symmetry and symmetry breaking . 51 6.4 The Ising ferromagnet in two dimensions . 52 6.5 Extreme Gibbs measures . 57 6.6 Uniqueness . 58 6.7 Ergodicity . 60 7 A variational characterisation of Gibbs measures 62 8 Large deviations theory 68 8.1 Motivation . 68 8.2 Definition . 70 8.3 Some results for Gibbs measures . 72 i 9 Models 73 9.1 Lattice Gases . 74 9.2 Magnetic Models . 75 9.3 Curie-Weiss model . 77 9.4 Continuous Ising model .