A Novel Splice Site Mutation in WAS Gene in Patient with Wiskott-Aldrich
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

Benign Tumors and Tumor-Like Lesions of the Vulva
Please do not remove this page Benign Tumors and Tumor-like Lesions of the Vulva Heller, Debra https://scholarship.libraries.rutgers.edu/discovery/delivery/01RUT_INST:ResearchRepository/12643402930004646?l#13643525330004646 Heller, D. (2015). Benign Tumors and Tumor-like Lesions of the Vulva. In Clinical Obstetrics & Gynecology (Vol. 58, Issue 3, pp. 526–535). Rutgers University. https://doi.org/10.7282/T3RN3B2N This work is protected by copyright. You are free to use this resource, with proper attribution, for research and educational purposes. Other uses, such as reproduction or publication, may require the permission of the copyright holder. Downloaded On 2021/09/23 14:56:57 -0400 Heller DS Benign Tumors and Tumor-like lesions of the Vulva Debra S. Heller, MD From the Department of Pathology & Laboratory Medicine, Rutgers-New Jersey Medical School, Newark, NJ Address Correspondence to: Debra S. Heller, MD Dept of Pathology-UH/E158 Rutgers-New Jersey Medical School 185 South Orange Ave Newark, NJ, 07103 Tel 973-972-0751 Fax 973-972-5724 [email protected] Funding: None Disclosures: None 1 Heller DS Abstract: A variety of mass lesions may affect the vulva. These may be non-neoplastic, or represent benign or malignant neoplasms. A review of benign mass lesions and neoplasms of the vulva is presented. Key words: Vulvar neoplasms, vulvar diseases, vulva 2 Heller DS Introduction: A variety of mass lesions may affect the vulva. These may be non-neoplastic, or represent benign or malignant neoplasms. Often an excision is required for both diagnosis and therapy. A review of the more commonly encountered non-neoplastic mass lesions and benign neoplasms of the vulva is presented. -
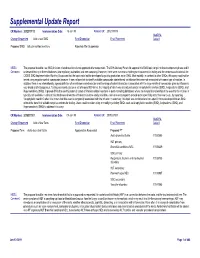
Detail Report
Supplemental Update Report CR Number: 2012319113 Implementation Date: 16-Jan-19 Related CR: 2012319113 MedDRA Change Requested Add a new SMQ Final Disposition Final Placement Code # Proposed SMQ Infusion related reactions Rejected After Suspension MSSO The proposal to add a new SMQ Infusion related reactions is not approved after suspension. The ICH Advisory Panel did approve this SMQ topic to go into the development phase and it Comment: underwent testing in three databases (two regulatory authorities and one company). However, there were numerous challenges encountered in testing and the consensus decision of the CIOMS SMQ Implementation Working Group was that the topic could not be developed to go into production as an SMQ. Most notably, in contrast to other SMQs, this query could not be tested using negative control compounds because it was not possible to identify suitable compounds administered via infusion that were not associated with some type of reaction. In addition, there is no internationally agreed definition of an infusion related reaction and the range of potential reactions associated with the large variety of compounds given by infusion is very broad and heterogenous. Testing was conducted on a set of around 500 terms, the majority of which was already included in Anaphylactic reaction (SMQ), Angioedema (SMQ), and Hypersensitivity (SMQ). It proved difficult to identify potential cases of infusion related reactions in post-marketing databases where the temporal relationship of the event to the infusion is typically not available. In clinical trial databases where this information is more easily available, users are encouraged to provide more specificity about the event, e.g., by reporting “Anaphylactic reaction” when it is known that this event is temporally associated with the infusion. -

Mouse Wipf1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Wipf1 Knockout Project (CRISPR/Cas9) Objective: To create a Wipf1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Wipf1 gene (NCBI Reference Sequence: NM_153138 ; Ensembl: ENSMUSG00000075284 ) is located on Mouse chromosome 2. 8 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 8 (Transcript: ENSMUST00000094681). Exon 3~6 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous mutants have immunological abnormalities, although lymphocyte development appears normal. Mutants show abnormal B and T cell proliferative responses, high serum immunoglobulin levels and impaired immunological synapse formation. Exon 3 starts from about 3.52% of the coding region. Exon 3~6 covers 85.26% of the coding region. The size of effective KO region: ~9629 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 5 6 8 Legends Exon of mouse Wipf1 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 3 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

The Guanine Nucleotide Exchange Factor Arhgef5 Plays Crucial Roles in Src-Induced Podosome Formation
1726 Research Article The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation Miho Kuroiwa, Chitose Oneyama, Shigeyuki Nada and Masato Okada* Department of Oncogene Research, Research institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan *Author for correspondence ([email protected]) Accepted 19 January 2011 Journal of Cell Science 124, 1726-1738 © 2011. Published by The Company of Biologists Ltd doi:10.1242/jcs.080291 Summary Podosomes and invadopodia are actin-rich membrane protrusions that play a crucial role in cell adhesion and migration, and extracellular matrix remodeling in normal and cancer cells. The formation of podosomes and invadopodia is promoted by upregulation of some oncogenic molecules and is closely related to the invasive potential of cancer cells. However, the molecular mechanisms underlying the podosome and invadopodium formation still remain unclear. Here, we show that a guanine nucleotide exchange factor (GEF) for Rho family GTPases (Arhgef5) is crucial for Src-induced podosome formation. Using an inducible system for Src activation, we found that Src-induced podosome formation depends upon the Src SH3 domain, and identified Arhgef5 as a Src SH3-binding protein. RNA interference (RNAi)-mediated depletion of Arhgef5 caused robust inhibition of Src-dependent podosome formation. Overexpression of Arhgef5 promoted actin stress fiber remodeling through activating RhoA, and the activation of RhoA or Cdc42 was required for Src-induced podosome formation. Arhgef5 was tyrosine-phosphorylated by Src and bound to Src to positively regulate its activity. Furthermore, the pleckstrin homology (PH) domain of Arhgef5 was required for podosome formation, and Arhgef5 formed a ternary complex with Src and phosphoinositide 3-kinase when Src and/or Arhgef5 were upregulated. -

Microrna Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton
cells Review MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton , , Karen Uray * y , Evelin Major and Beata Lontay * y Department of Medical Chemistry, Faculty of Medicine, University of Debrecen, 4032 Debrecen, Hungary; [email protected] * Correspondence: [email protected] (K.U.); [email protected] (B.L.); Tel.: +36-52-412345 (K.U. & B.L.) The authors contributed equally to the manuscript. y Received: 11 June 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: MicroRNAs (miRNAs) are key modulators of post-transcriptional gene regulation in a plethora of processes, including actin–myosin cytoskeleton dynamics. Recent evidence points to the widespread effects of miRNAs on actin–myosin cytoskeleton dynamics, either directly on the expression of actin and myosin genes or indirectly on the diverse signaling cascades modulating cytoskeletal arrangement. Furthermore, studies from various human models indicate that miRNAs contribute to the development of various human disorders. The potentially huge impact of miRNA-based mechanisms on cytoskeletal elements is just starting to be recognized. In this review, we summarize recent knowledge about the importance of microRNA modulation of the actin–myosin cytoskeleton affecting physiological processes, including cardiovascular function, hematopoiesis, podocyte physiology, and osteogenesis. Keywords: miRNA; actin; myosin; actin–myosin complex; Rho kinase; cancer; smooth muscle; hematopoiesis; stress fiber; gene expression; cardiovascular system; striated muscle; muscle cell differentiation; therapy 1. Introduction Actin–myosin interactions are the primary source of force generation in mammalian cells. Actin forms a cytoskeletal network and the myosin motor proteins pull actin filaments to produce contractile force. All eukaryotic cells contain an actin–myosin network inferring contractile properties to these cells. -

Surgical Excision of Eyelid Lesions Reference Number: CP.VP.75 Coding Implications Last Review Date: 12/2020 Revision Log
Clinical Policy: Surgical Excision of Eyelid Lesions Reference Number: CP.VP.75 Coding Implications Last Review Date: 12/2020 Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description: The majority of eyelid lesions are benign, ranging from innocuous cysts and chalazion/hordeolum to nevi and papillomas. Key features that should prompt further investigation include gradual enlargement, central ulceration or induration, irregular borders, eyelid margin destruction or loss of lashes, and telangiectasia. This policy describes the medical necessity requirements for surgical excision of eyelid lesions. Policy/Criteria I. It is the policy of health plans affiliated with Centene Corporation® (Centene) that surgical excision and repair of eyelid or conjunctiva due to lesion or cyst or eyelid foreign body removal is medically necessary for any of the following indications: A. Lesion with one or more of the following characteristics: 1. Bleeding; 2. Persistent or intense itching; 3. Pain; 4. Inflammation; 5. Restricts vision or eyelid function; 6. Misdirects eyelashes or eyelid; 7. Displaces lacrimal puncta or interferes with tear flow; 8. Touches globe; 9. Unknown etiology with potential for malignancy; B. Lesions classified as one of the following: 1. Malignant; 2. Benign; 3. Cutaneous papilloma; 4. Cysts; 5. Embedded foreign bodies; C. Periocular warts associated with chronic conjunctivitis. Background The majority of eyelid lesions are benign, ranging from innocuous cysts and chalazion/hordeolum to nevi and papillomas. Key features that should prompt further investigation include gradual enlargement, central ulceration or induration, irregular borders, eyelid margin destruction or loss of lashes, and telangiectasia. Benign tumors, even though benign, often require removal and therefore must be examined carefully and the differential diagnosis of a malignant eyelid tumor considered and the method of removal planned. -

2016 Essentials of Dermatopathology Slide Library Handout Book
2016 Essentials of Dermatopathology Slide Library Handout Book April 8-10, 2016 JW Marriott Houston Downtown Houston, TX USA CASE #01 -- SLIDE #01 Diagnosis: Nodular fasciitis Case Summary: 12 year old male with a rapidly growing temple mass. Present for 4 weeks. Nodular fasciitis is a self-limited pseudosarcomatous proliferation that may cause clinical alarm due to its rapid growth. It is most common in young adults but occurs across a wide age range. This lesion is typically 3-5 cm and composed of bland fibroblasts and myofibroblasts without significant cytologic atypia arranged in a loose storiform pattern with areas of extravasated red blood cells. Mitoses may be numerous, but atypical mitotic figures are absent. Nodular fasciitis is a benign process, and recurrence is very rare (1%). Recent work has shown that the MYH9-USP6 gene fusion is present in approximately 90% of cases, and molecular techniques to show USP6 gene rearrangement may be a helpful ancillary tool in difficult cases or on small biopsy samples. Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors, 5th edition. Mosby Elsevier. 2008. Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, Ye Y, Lau AW, Wang X, Oliveira AM. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011 Oct;91(10):1427-33. Amary MF, Ye H, Berisha F, Tirabosco R, Presneau N, Flanagan AM. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013 Jul;463(1):97-8. CONTRIBUTED BY KAREN FRITCHIE, MD 1 CASE #02 -- SLIDE #02 Diagnosis: Cellular fibrous histiocytoma Case Summary: 12 year old female with wrist mass. -

Genetic Heterogeneity Intuberous Sclerosis: Phenotypic Correlations
J Med Genet: first published as 10.1136/jmg.27.7.418 on 1 July 1990. Downloaded from 4184 Med Genet 1990; 27: 418-421 Genetic heterogeneity in tuberous sclerosis: phenotypic correlations I M Winship, J M Connor, P H Beighton Abstract sistently present in families in whom the gene for There is increasing evidence for genetic hetero- TSC is not on 9q34. We conclude that confetti geneity in tuberous sclerosis (TSC) on the basis of depigmentation and nuchal skin tags may be clinical linkage analysis in affected kindreds. We have per- pointers to an alternative locus for TSC. formed a detailed assessment of an affected South African family in which there is no evidence of linkage to chromosome 9 markers. The affected persons have atypical clinical features, namely Tuberous sclerosis (TSC) is inherited as an autosomal prominent nuchal skin tags, a confetti pattern of dominant trait and is characterised by multisystem hypopigmentation of the skin of the lower legs, and hamartosis. The areas of predilection are the skin, absence of ungual fibromata. Further investigation central nervous system, kidneys, and heart, while of these unusual phenotypic features is warranted in other organs are less frequently affected.' Certain skin order to determine whether these lesions are con- lesions are pathognomonic of TSC (adenoma seba- ceum, periungual fibromata, shagreen patches, fibrous facial plaques). Other skin changes may be copyright. MRC Unit for Inherited Skeletal Disorders, Department suggestive (ash leaf macules) or compatible with the of Human Genetics, University of Cape Town Medical diagnosis of TSC in the appropriate clinical setting School, Observatory 7925, South Africa. -

SUPPLEMENTARY APPENDIX a Homozygous Missense Variant in UBE2T Is Associated with a Mild Fanconi Anemia Phenotype
SUPPLEMENTARY APPENDIX A homozygous missense variant in UBE2T is associated with a mild Fanconi anemia phenotype Laura Schultz-Rogers, 1* Francis P. Lach, 2* Kimberly A. Rickman, 2 Alejandro Ferrer, 1 Abhishek A. Mangaonkar, 3 Tanya L. Schwab, 4 Christo - pher T. Schmitz, 4 Karl J. Clark, 4 Nikita R. Dsouza, 5 Michael T. Zimmermann, 5,6 Mark Litzow, 3 Nicole Jacobi, 7 Eric W. Klee, 1,8 Agata Smogorzewska 2# and Mrinal M. Patnaik 3# 1Center for Individualized Medicine, Mayo Clinic, Rochester, MN; 2Laboratory of Genome Maintenance, The Rockefeller University, New York, NY; 3De - partment of Hematology, Mayo Clinic, Rochester, MN; 4Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN; 5Bioinformatics Re - search and Development Laboratory, Genomics Sciences and Precision Medicine Center, Medical College of Wisconsin, Milwaukee, WI; 6Clinical and Translational Sciences Institute, Medical College of Wisconsin, Milwaukee, WI; 7Department of Hematology Oncology, Hennepin County Medical Center, Min - neapolis, MN and 8Department of Clinical Genomics, Mayo Clinic, Rochester, MN, USA *LS-R and FPL contributed equally as co-first authors. #EWK, AS and MMP contributed equally as co-senior authors. Correspondence: MRINAL PATNAIK - [email protected] AGATA SMOGORZEWSKA - [email protected] doi:10.3324/haematol.2020.259275 Supplemental Information Homozygous missense variant in UBE2T is associated with mild Fanconi anemia phenotype Laura Schultz-Rogers1*, Francis P. Lach2*, Kimberly A. Rickman2, Alejandro Ferrer1,