Preparation of N-‐(Boc)-‐Allylglycine Methyl Ester
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

Metabolic and Homeostatic Changes In
University of Dundee Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy-Mitochondria, Calcium Dynamics and Reactive Oxygen Species Kovac, Stjepana; Dinkova Kostova, Albena T; Herrmann, Alexander M; Melzer, Nico; Meuth, Sven G; Gorji, Ali Published in: International Journal of Molecular Sciences DOI: 10.3390/ijms18091935 Publication date: 2017 Licence: CC BY Document Version Publisher's PDF, also known as Version of record Link to publication in Discovery Research Portal Citation for published version (APA): Kovac, S., Dinkova Kostova, A. T., Herrmann, A. M., Melzer, N., Meuth, S. G., & Gorji, A. (2017). Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy-Mitochondria, Calcium Dynamics and Reactive Oxygen Species. International Journal of Molecular Sciences, 18(9), 1-19. [1935]. https://doi.org/10.3390/ijms18091935 General rights Copyright and moral rights for the publications made accessible in Discovery Research Portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from Discovery Research Portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 29. Sep. 2021 International Journal of Molecular Sciences Review Metabolic and Homeostatic Changes in Seizures and Acquired Epilepsy—Mitochondria, Calcium Dynamics and Reactive Oxygen Species Stjepana Kovac 1,*, Albena T. -

Ratio of Phosphate to Amino Acids
National Institute for Health and Care Excellence Final Neonatal parenteral nutrition [D10] Ratio of phosphate to amino acids NICE guideline NG154 Evidence reviews February 2020 Final These evidence reviews were developed by the National Guideline Alliance which is part of the Royal College of Obstetricians and Gynaecologists FINAL Error! No text of specified style in document. Disclaimer The recommendations in this guideline represent the view of NICE, arrived at after careful consideration of the evidence available. When exercising their judgement, professionals are expected to take this guideline fully into account, alongside the individual needs, preferences and values of their patients or service users. The recommendations in this guideline are not mandatory and the guideline does not override the responsibility of healthcare professionals to make decisions appropriate to the circumstances of the individual patient, in consultation with the patient and/or their carer or guardian. Local commissioners and/or providers have a responsibility to enable the guideline to be applied when individual health professionals and their patients or service users wish to use it. They should do so in the context of local and national priorities for funding and developing services, and in light of their duties to have due regard to the need to eliminate unlawful discrimination, to advance equality of opportunity and to reduce health inequalities. Nothing in this guideline should be interpreted in a way that would be inconsistent with compliance with those duties. NICE guidelines cover health and care in England. Decisions on how they apply in other UK countries are made by ministers in the Welsh Government, Scottish Government, and Northern Ireland Executive. -

Pharmacological Studies on a Locust Neuromuscular Preparation
J. Exp. Biol. (1974). 6i, 421-442 421 *&ith 2 figures in Great Britain PHARMACOLOGICAL STUDIES ON A LOCUST NEUROMUSCULAR PREPARATION BY A. N. CLEMENTS AND T. E. MAY Woodstock Research Centre, Shell Research Limited, Sittingbourne, Kent {Received 13 March 1974) SUMMARY 1. The structure-activity relationships of agonists of the locust excitatory neuromuscular synapse have been reinvestigated, paying particular attention to the purity of compounds, and to the characteristics and repeatability of the muscle response. The concentrations of compounds required to stimu- late contractions of the retractor unguis muscle equal in force to the neurally evoked contractions provided a measure of the relative potencies. 2. Seven amino acids were capable of stimulating twitch contractions, glutamic acid being the most active, the others being analogues or derivatives of glutamic or aspartic acid. Aspartic acid itself had no excitatory activity. 3. Excitatory activity requires possession of two acidic groups, separated by two or three carbon atoms, and an amino group a to a carboxyl. An L-configuration appears essential. The w-acidic group may be a carboxyl, sulphinyl or sulphonyl group. Substitution of any of the functional groups generally causes total loss of excitatory activity, but an exception is found in kainic acid in which the nitrogen atom forms part of a ring. 4. The investigation of a wide variety of compounds revealed neuro- muscular blocking activity among isoxazoles, hydroxylamines, indolealkyl- amines, /?-carbolines, phenazines and phenothiazines. No specific antagonist of the locust glutamate receptor was found, but synaptic blocking agents of moderately high activity are reported. INTRODUCTION The study of arthropod neuromuscular physiology has been impeded by the lack of an antagonist which can be used to block excitatory synaptic transmission by a specific postsynaptic effect. -

Blockade of Photically Induced Epilepsy by 'Dopamine Agonist' Ergot Alkaloids
Psychopharmacology 57, 57-62 (1978) Psychopharmacology © by Springer-Verlag 1978 Blockade of Photically Induced Epilepsy by 'Dopamine Agonist' Ergot Alkaloids GILL ANLEZARK and BRIAN MELDRUM Department of Neurology, Institute of Psychiatry, De Crespigny Park, London, SES 8AF, United Kingdom Abstract. The effect of the intravenous administration of the visual evoked response in the lateral geniculate of ergot alkaloids on epileptic responses to intermittent body or occipital cortex correlates well with the reduc- photic stimulation (IPS) has been studied in adolescent tion in photosensitivity (Vuillon-Cacciuttolo et aI., baboons, Papio papio, from Senegal. Ergocornine, 1973). In addition to the evidence for serotonin 1- 2 mgjkg, produced marked autonomic and be- agonist and antagonist actions of ergot alkaloids in havioural effects, slowed the EEG, and abolished the CNS, it has recently been demonstrated that some myoclonic responses to IPS for 30-90 min. Ergo- ergot derivatives interact with cerebral dopamine metrine, 1 mgjkg, activated the EEG and blocked the (DA) receptors. Thus several ergot alkaloids stimulate induction of myoclonic responses for 1- 3 h. Bromo- dopamine-sensitive adenylate cyclase (von Hungen criptine, 0.5-4 mgjkg, did not consistently prevent et aI., 1974; da Prada et aI., 1975) and modify motor myoclonic responses to IPS. After pretreatment with activity in intact or brain-lesioned rats in ways a subconvulsant dose of allylglycine (180- 200 mgjkg), indicative of dopaminergic activation (see Discus- lysergic acid diethylamide, 0.1 mgjkg, retained the sion). capacity to block myoclonic responses to IPS, and We previously reported that the dopamine agonist ergocornine 1 mgjkg reduced such responses. The apomorphine reduces photically induced epilepsy in convulsant effect of allylglycine was enhanced, how- Papio papio (Meldrum et aI., 1975a) and that ergot ever, so that prolonged seizure sequences began alkaloids which cause contralateral turning in mice 19- 96 min after ergocornine administration. -

The Antimyoclonic Action of Clonazepam Through a Gaba-Independent Mechanism
Ind. J. Physiol. Pharmac., Volume SS, Number 4, 1989 SHORT COMMUNICATION THE ANTIMYOCLONIC ACTION OF CLONAZEPAM THROUGH A GABA-INDEPENDENT MECHANISM VANAJA PAUL· AND M. S. KRISHNAMOORTHY Department ofPharmacology and Environmental Toxicology, Dr. A. L. M. Postgraduate Institute ofBasic Medical Sciences, University of Madras, Taramani, Madras - 600 113 ( Received on August 28, 1989) Abstract: The present Itudy investigatea whether clonazepam exerts itl antimyoclonic action through a GABA independent mechanism. We have Itudied the antimyoclonic effect of clona zepam and compared it with that of aminooxyacetic acid (AOAA), a GABA transaminase inhibitor, against myoclonul induced by picrotoxin, a GABA receptor antagonilt and aIlylglycine, a drug which inhibits synthesis and release of GABA. We have also iavestigated the effect of c10nazepam against picrotoxin-induced myodonus in rats pretreated with either AOAA or aubmyoclonic dOle of ahylgylycine. Clonazepam pretreatment inhibited both picrotoxin and allylglycine-induced myoeionul whereal AOAA was effective in inhibiting only picrotoxin-induced myoclonul. The protective effect of clonazepam against picrotoxin-induced myoclonus was potentiated by AOAA pretreatment. Moreover, cloDazepam afforded protection againlt picrotoxin-induced myoclonus in rats pretreated with a submyoc1onic GABA reducing dose of allylglycine. Thele findings indicate that a GABA independent mechanilm may also be involved in the antimyodonic action of c1onazepam. Key worde : clonazepam picrotoxin allylglycine aminooxyacetic acid INTRODUCTION induced by picrotoxin, a GABA receptor antagonist . (6, 7) and· that produced by allylglycine, a drug Clinical (1. 2) and experimental (3) findings which inhibits synthesis (8) and release (9) of GABA. reveal that myoclonic syndrome is causally related In another set of experiments we have investigated to an impairment of gamma aminobutyric acid the antimyoclonic potency of clonazepam against (GABA) activity in the brain. -

WO 2017/092759 Al 8 June 2017 (08.06.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/092759 Al 8 June 2017 (08.06.2017) P O P C T (51) International Patent Classification: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 38/00 (2006.01) C07K 14/705 (2006.01) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/DK2016/050369 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, 16 November 2016 (16.1 1.2016) TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, PA 2015 70783 1 December 2015 (01 .12.2015) DK TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: UNIVERSITY OF COPENHAGEN DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, [DK/DK]; N0rregade 10, 1165 Copenhagen K (DK). -
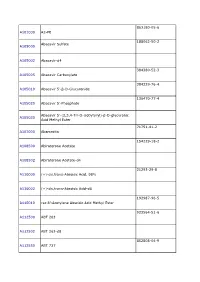
TRC Price List 2010
863180-05-6 A101000 A2-PE 188062-50-2 Abacavir Sulfate A105000 A105002 Abacavir-d4 384380-52-3 A105005 Abacavir Carboxylate 384329-76-4 A105010 Abacavir 5’-β-D-Glucuronide 136470-77-4 A105020 Abacavir 5’-Phosphate Abacavir 5’-(2,3,4-Tri-O-isobytyryl)-β-D-glucuronic A105030 Acid Methyl Ester 71751-41-2 A107000 Abamectin 154229-18-2 A108500 Abiraterone Acetate A108502 Abiraterone Acetate-d4 21293-29-8 A110000 (+)-cis,trans-Abscisic Acid, 98% A110002 (+)-cis,trans-Abscisic Acid-d6 192987-96-5 A110010 rac 8’-Acetylene Abscisic Acid Methyl Ester 923564-51-6 A112500 ABT 263 A112502 ABT 263-d8 852808-04-9 A112550 ABT 737 A112552 ABT 737-d8 912444-00-9 A112580 ABT 888 447407-36-5 A115600 AC 42 77337-73-6 A120000 Acamprosate Calcium A120002 Acamprosate-d12 Calcium Trihydrate A120003 Acamprosate-d6 Calcium Trihydrate 56180-94-0 A123500 Acarbose A123502 Acarbose-d4 117065-98-2 A123505 Acarbose Tridecaacetate 34381-68-5 A123800 Acebutolol Hydrochloride Acebutolol 37517-30-9 A123801 Discontinued. -

Effects of Reversible Inactivation of the Dorsomedial Hypothalamus on Panic- and Anxiety-Related Responses in Rats J.O.G
Brazilian Journal of Medical and Biological Research Online Provisional Version ISSN 0100-879X This Provisional PDF corresponds to the article as it appeared upon acceptance. Fully formatted PDF and full text (HTML) versions will be made available soon. Effects of reversible inactivation of the dorsomedial hypothalamus on panic- and anxiety-related responses in rats J.O.G. Nascimento1, H. Zangrossi Jr.2 and M.B. Viana3 1Departamento de Psiquiatria, Universidade Federal de São Paulo, São Paulo, SP, Brasil 2Departamento de Farmacologia, Faculdade de Medicina de Ribeirao Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil 3Departamento de Biociências, Universidade Federal de São Paulo, Santos, SP, Brasil Abstract The medial hypothalamus is part of a neurobiological substrate controlling defensive behavior. It has been shown that a hypo- thalamic nucleus, the dorsomedial hypothalamus (DMH), is involved in the regulation of escape, a defensive behavior related to panic attacks. The role played by the DMH in the organization of conditioned fear responses, however, is less clear. In the present study, we investigated the effects of reversible inactivation of the DMH with the GABAA agonist muscimol on inhibitory avoidance acquisition and escape expression by male Wistar rats (approximately 280 g in weight) tested in the elevated T-maze (ETM). In the ETM, inhibitory avoidance, a conditioned defensive response, has been associated with generalized anxiety dis- order. Results showed that intra-DMH administration of the GABAA receptor agonist muscimol inhibited escape performance, suggesting an antipanic-like effect (P < 0.05), without changing inhibitory avoidance acquisition. Although a higher dose of muscimol (1.0 nmol/0.2 µL; N = 7) also altered locomotor activity in an open field when compared to control animals (0.2 µL saline; N = 13) (P < 0.05), the lower dose (0.5 nmol/0.2 µL; N = 12) of muscimol did not cause any motor impairment. -

Psychopharmacology of Memory Modulation
BEHAVIOURAL BRAIN RESEARCH ELSEVIER Behavioural Brain Research 77 (1996) 1-21 Review article Psychopharmacology of memory modulation: Evidence for multiple interaction among neurotransmitters and hormones Claudio Castellano a.,, Simona Cabib ", Stefano Puglisi-Allegra b " Istituto di Psicobiologia e Psicofarmacologia (CNR), via Reno 1, Rome 1-00198, Italy b Department of Psychology (Neuroscience section), University of Rome 'La Sapienza', Rome, Italy Received 16 January 1995; revised 26 June 1995; accepted 26 June 1995 Abstract Experimental results are reviewed which indicate that memory storage can be altered by a number of post-training treatments that affect different hormones and neurotransmitters. Moreover, evidence was reported which suggests that the action of treatments effective on memory processes involves interactions among different systems, consistently with the complexity of brain systems. In the last decade, inbred strains have been exploited to investigate the role of neurotransmitter and hormone systems in learning and memory, leading to behavioural and neurochemical correlations based on strain differences that provide unique information on the biological systems underlying behaviour. Research carded out on the inbred strains of mice C57BL/6 (C57) and DBA/2 (DBA), demonstrates that the genetic makeup plays an important role in modulating response to drug administration. Thus, recent results have shown that in C57 mice, similarly to what occurs in outbred strains of mice or in rats, GABAergic agonists impair memory and antagonists improve it, whilst the opposite is evident in the DBA strain. By contrast, post-training administration of selective Ell or D2 agonists impairs and post-training administration of selective antagonists improves retention in DBA mice, whilst these agents have opposite effects in the C57 strain. -

To My Parents the Secret of Science Is to Ask the Right Question, and It Is
To my parents The secret of science is to ask the right question, and it is the choice of problem more than anything else that marks a man of genius in the scientific world. Sir Heniy Tizard. There's an old saying in research: it's okay to sleep with a hypothesis, but you should never marry one. J. William Langston. ProQuest Number: U553556 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest U553556 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 ROLE OF DOPAMINE IN PILOCARPINE INDUCED MOTOR SEIZURES. A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy in the Faculty of Medicine, University of London. Presented by GHADA KHALIL AL-TAJIR (B.S c. H ons.) The School of Pharmacy, University of London, 29-39 Brunswick Square, London WC1N 1AX. May, 1992 II ACKNOWLEDGEMENTS I would like to express my gratitude to the Committee of Vice Chancellors and Principals for giving me the Overseas Research Studentship award which supported this work. -

Amino Acids and Peptides in Ball Milling Thomas-Xavier Métro, Evelina Colacino, Jean Martinez, Frédéric Lamaty
Amino Acids and Peptides in Ball Milling Thomas-Xavier Métro, Evelina Colacino, Jean Martinez, Frédéric Lamaty To cite this version: Thomas-Xavier Métro, Evelina Colacino, Jean Martinez, Frédéric Lamaty. Amino Acids and Pep- tides in Ball Milling. Achim Stolle; Brindaban Ranu. Ball Milling Towards Green Synthesis: Ap- plications, Projects, Challenges, Royal Society of Chemistry, pp.114-150, 2014, 978-1-78262-348-9. 10.1039/9781782621980-00114. hal-02364140 HAL Id: hal-02364140 https://hal.archives-ouvertes.fr/hal-02364140 Submitted on 25 May 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. CHAPTER 6 Amino Acids and Peptides in Ball Millingy THOMAS-XAVIER ME´TRO, EVELINA COLACINO, JEAN MARTINEZ AND FRE´DE´RIC LAMATY* Institut des Biomole´cules Max Mousseron, UMR 5247 CNRS–UM1–UM2– ENSCM, Universite´ Montpellier 2, Place E. Bataillon, 34095 Montpellier Cedex 5, France *Email: [email protected] 6.1 Introduction For many years, pharmaceutical companies have focused their attention on the development of drugs based on the biological activity of small molecules. More recently, peptides have been recognized as efficient active pharma- ceutical ingredients and new delivery systems have moved them forward in the modern therapeutic arsenal.