Microarray Analysis Clc5 Wt and Ko Mice, 8Weeks Old, Male, 10
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prox1regulates the Subtype-Specific Development of Caudal Ganglionic
The Journal of Neuroscience, September 16, 2015 • 35(37):12869–12889 • 12869 Development/Plasticity/Repair Prox1 Regulates the Subtype-Specific Development of Caudal Ganglionic Eminence-Derived GABAergic Cortical Interneurons X Goichi Miyoshi,1 Allison Young,1 Timothy Petros,1 Theofanis Karayannis,1 Melissa McKenzie Chang,1 Alfonso Lavado,2 Tomohiko Iwano,3 Miho Nakajima,4 Hiroki Taniguchi,5 Z. Josh Huang,5 XNathaniel Heintz,4 Guillermo Oliver,2 Fumio Matsuzaki,3 Robert P. Machold,1 and Gord Fishell1 1Department of Neuroscience and Physiology, NYU Neuroscience Institute, Smilow Research Center, New York University School of Medicine, New York, New York 10016, 2Department of Genetics & Tumor Cell Biology, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, 3Laboratory for Cell Asymmetry, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan, 4Laboratory of Molecular Biology, Howard Hughes Medical Institute, GENSAT Project, The Rockefeller University, New York, New York 10065, and 5Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724 Neurogliaform (RELNϩ) and bipolar (VIPϩ) GABAergic interneurons of the mammalian cerebral cortex provide critical inhibition locally within the superficial layers. While these subtypes are known to originate from the embryonic caudal ganglionic eminence (CGE), the specific genetic programs that direct their positioning, maturation, and integration into the cortical network have not been eluci- dated. Here, we report that in mice expression of the transcription factor Prox1 is selectively maintained in postmitotic CGE-derived cortical interneuron precursors and that loss of Prox1 impairs the integration of these cells into superficial layers. Moreover, Prox1 differentially regulates the postnatal maturation of each specific subtype originating from the CGE (RELN, Calb2/VIP, and VIP). -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Gene List of the Targeted NGS MCD and CCA Gene Panel AKT3,ALX1
Gene List of the targeted NGS MCD and CCA gene panel AKT3,ALX1,ALX3,ALX4,AMPD2,ARFGEF2,ARID1B,ARX,ASPM,ATR,ATRX,B3GALTL,BRPF1,c12orf57,C6orf70,CASK,CCND2,CDK5RAP2,CDON,C ENPJ,CEP170,CHMP1A,COL4A1,CREBBP,CYP11A1,DCHS1,DCLK1,DCX,DHCR24,DHCR7,DIS3L2,DISC1,DISP1,DLL1,DMRTA2,DYNC1H1,DYRK1 A,EARS2,EFNB1,EMX1,EOMES,EP300,ERBB4,ERMARD,EXOSC3,FAM36A,FGF8,FGFR1,FGFR2,FLNA,FOXC1,FOXG1,FOXH1,FZD10,GLI2,GLI3,GP R56,GPSM2,HCCS,HESX1,HNRNPU,IGBP1,IGFBP1,ISPD,ITPA,KAL1,KAT6B,KATNB1,KIAA1279,KIF14,KIF1A,KIF1B,KIF21A,KIF2A,KIF5C,KIF7,L1 CAM,LAMB1,LAMC3,LRP2,MCPH1,MED12,MID1,NDE1,NFIB,NPC1,NR2F1,NSD1,NTRK1,NTRK3,OCEL1,OPA1,OTX2,PAFAH1B1,PAX6,PEX1,PHF1 0,PIK3R2,POLR3A,POLR3B,POMT1,POMT2,PTCH1,PTPRS,PYCR1,RAB3GAP1,RARS2,RELN,RFX3,ROBO1,ROBO3,RPS6KA3,RTTN,SATB2,SEPSEC S,SHH,SIX3,SLC12A6,SOX2,SPOCK1,SRPX2,TBCD,TBCE,TCF4,TDGF1,TEAD1,THBS2,TMEM5,TSC1,TSC2,TSEN15,TSEN2,TSEN34,TSEN54,TUBA1 A,TUBA8,TUBB,TUBB2A,TUBB2B,TUBB3,TUBB4A,TUBG1,VAX1,VRK1,WDR47,WDR62,ZBTB18,ZEB2,ZIC2. Gene List of the targeted NGS epilepsy gene panel AARS, ADGRV1, ADRA2B, ADSL, ALDH4A1, ALDH7A1, ALG13, ALPL, ARHGEF15, ARHGEF9, ARX, ASAH1, ATP1A2, ATP1A3, BRD2, CACNA1A, CACNA1H, CACNA2D2, CACNB4, CBL, CDKL5, CERS1, CHD2, CHRNA2, CHRNA4, CHRNB2, CLCN2, CLCN4, CLN8, CLTC, CNKSR2, CNTNAP2, CPA6, CPLX1, CSNK1G1, CSNK2B, CTNND2, DEPDC5, DHDDS, DNM1, DOCK7, DYNC1H1, EEF1A2, EFHC1, EIF2S3, EMC1, EPM2A, FASN, FLNA, FOXG1, GABBR2, GABRA1, GABRA2, GABRA3, GABRB2, GABRB3, GABRD, GABRG2, GAL, GNAO1, GOSR2, GRIA1, GRIN1, GRIN2A, GRIN2B, HCN1, HCN4, HDAC4, HNRNPU, IDH3A, IQSEC2, JRK, KCNA1, KCNA2, KCNB1, -

Calcium Channel Diversity (Updated 2003)
Calcium channel diversity (updated 2003) Annette C Dolphin Department of Pharmacology University College London Gower St. London WC1E6BT, UK tel. +44 207 679 3276 fax +44 207 813 2808 [email protected] 2 I Calcium channel diversity III Contents list Introduction 1. Low threshold and high threshold voltage-gated calcium channels 2. Structure-function studies of voltage-gated calcium channels 3. The CaV1 or SCDF family of calcium channels 4. The CaV2 or ABE family of calcium channels 5. The CaV3 or GHI family of calcium channels 6. Calcium channel subunits are localised to different parts of the neuron 7. Intracellular calcium channels IV Article definition Calcium channels, when open, allow Ca2+ ions to pass down their electrochemical gradient. Here I describe the subtypes of Ca2+ channel and their functions V Introduction Voltage-dependent calcium channels (VDCCs) were first identified in crustacean muscle by Paul Fatt and Bernard Katz (1953) and subsequently studied extensively by Hagiwara and colleagues. These muscles still showed action potentials in the absence of external Na+, that were dependent on calcium (Ca2+) entry. Ca2+ is generally present at a concentration of a few mM in the extracellular space, but inside the cell, the cytoplasmic concentration is about 0.1µM, kept low by a number of different pumps and buffering systems, as well as the general impermeability of the plasma membrane to the entry of Ca2+. VDCCs have subsequently been found in all types of excitable cell in vertebrates and invertebrates, and even plants. They fulfil numerous functions depending on the tissue, and it is thus not surprising that a number of subclasses of VDCC have been identified. -

Identifying Nootropic Drug Targets Via Large-Scale Cognitive GWAS and Transcriptomics
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.06.934752; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Title: Identifying Nootropic Drug Targets via Large-Scale Cognitive GWAS and Transcriptomics Max Lam1, 2, 3,4, Chia-Yen, Chen3,5,6, Xia Yan7,8, W. David Hill9, 10, Joey W. Trampush11, Jin Yu1, Emma Knowles12,13,14, Gail Davies9, 10, Eli Stahl15, 16, Laura Huckins15, 16, David C. Liewald10, Srdjan Djurovic17, 18, Ingrid Melle18, 19, Andrea Christoforou20, Ivar Reinvang21, Pamela DeRosse1, 22, 23, Astri J. Lundervold24, Vidar M. Steen18, 20, Thomas Espeseth19, 21, Katri Räikkönen25, Elisabeth Widen26, Aarno Palotie26, 27, 28, Johan G. Eriksson29, 30, 31, Ina Giegling32, Bettina Konte32, Annette M. Hartmann32, Panos Roussos15, 16, 33, Stella Giakoumaki34, Katherine E. Burdick15, 33, 35, Antony Payton36, William Ollier37, 38, Ornit Chiba- Falek39, Deborah K. Koltai39, 40 , Anna C. Need41, Elizabeth T. Cirulli42, Aristotle N. Voineskos43, Nikos C. Stefanis44, 45, 46, Dimitrios Avramopoulos47, 48, Alex Hatzimanolis44, 45, 46, Nikolaos Smyrnis44, 45, Robert M. Bilder49, Nelson A. Freimer49, Tyrone D. Cannon50, 51, Edythe London49, Russell A. Poldrack52, Fred W. Sabb53, Eliza Congdon49, Emily Drabant Conley54, Matthew A. Scult55, Dwight Dickinson56, Richard E. Straub57, Gary Donohoe58, Derek Morris58, Aiden Corvin59, Michael Gill59, Ahmad R. Hariri55, Daniel R. Weinberger57, Neil Pendleton60, Panos Bitsios61, Dan Rujescu32, Jari Lahti25, 62, Stephanie Le Hellard18, 20, Matthew C. -

The L-Type Calcium Channel in the Heart: the Beat Goes On
The L-type calcium channel in the heart: the beat goes on Ilona Bodi, … , Shahab A. Akhter, Arnold Schwartz J Clin Invest. 2005;115(12):3306-3317. https://doi.org/10.1172/JCI27167. Review Sydney Ringer would be overwhelmed today by the implications of his simple experiment performed over 120 years ago showing that the heart would not beat in the absence of Ca2+. Fascination with the role of Ca2+ has proliferated into all aspects of our understanding of normal cardiac function and the progression of heart disease, including induction of cardiac hypertrophy, heart failure, and sudden death. This review examines the role of Ca2+ and the L-type voltage- dependent Ca2+ channels in cardiac disease. Find the latest version: https://jci.me/27167/pdf Review The L-type calcium channel in the heart: the beat goes on Ilona Bodi, Gabor Mikala, Sheryl E. Koch, Shahab A. Akhter, and Arnold Schwartz Institute of Molecular Pharmacology and Biophysics, University of Cincinnati College of Medicine, Department of Surgery, Cincinnati, Ohio, USA. Sydney Ringer would be overwhelmed today by the implications of his simple experiment performed over 120 years ago showing that the heart would not beat in the absence of Ca2+. Fascination with the role of Ca2+ has pro- liferated into all aspects of our understanding of normal cardiac function and the progression of heart disease, including induction of cardiac hypertrophy, heart failure, and sudden death. This review examines the role of Ca2+ and the L-type voltage-dependent Ca2+ channels in cardiac disease. When Sydney Ringer (1) discovered the vital role of Ca2+ in the exchanger (NCX). -
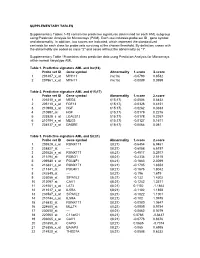
Supplementary Tables 1-18 Contain the Predictive Signatures Determined for Each AML Subgroup Using Prediction Analysis for Microarrays (PAM)
SUPPLEMENTARY TABLES Supplementary Tables 1-18 contain the predictive signatures determined for each AML subgroup using Prediction Analysis for Microarrays (PAM). Each row indicates probe set ID , gene symbol and abnormality. In addition, two scores are indicated, which represent the standardized centroids for each class for probe sets surviving at the chosen threshold. By definition, cases with the abnormality are coded as class "2" and cases without the abnormality as "1". Supplementary Table 19 contains class prediction data using Prediction Analysis for Microarrays within normal karyotype AML. Table 1. Predictive signature AML and inv(16) Probe set ID Gene symbol Abnormality 1.score 2.score 1 201497_x_at MYH11 inv(16) -0.0793 0.8532 2 207961_x_at MYH11 inv(16) -0.0089 0.0959 Table 2. Predictive signature AML and t(15;17) Probe set ID Gene symbol Abnormality 1.score 2.score 1 226210_s_at MEG3 t(15;17) -0.0506 0.6432 2 205110_s_at FGF13 t(15;17) -0.0326 0.4151 3 210998_s_at HGF t(15;17) -0.0262 0.3333 4 210997_at HGF t(15;17) -0.0179 0.2276 5 223828_s_at LGALS12 t(15;17) -0.0178 0.2267 6 210794_s_at MEG3 t(15;17) -0.0127 0.1611 7 204537_s_at GABRE t(15;17) -0.0064 0.081 Table 3. Predictive signature AML and t(8;21) Probe set ID Gene symbol Abnormality 1.score 2.score 1 205529_s_at RUNX1T1 t(8;21) -0.6454 6.9461 2 228827_at --- t(8;21) -0.6058 6.5197 3 205528_s_at RUNX1T1 t(8;21) -0.4917 5.2917 4 213194_at ROBO1 t(8;21) -0.2334 2.5115 5 206940_s_at POU4F1 t(8;21) -0.1883 2.0269 6 216831_s_at RUNX1T1 t(8;21) -0.1705 1.8353 7 211341_at -

ADX609 3301 Athena Newborndx Gene Panel Update 3-30-20.Indd
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 3- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9