Whole Genome Sequencing Analysis of Lung
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Co-Construction of Mine Villages—An Important Way for Coal Mining Enterprises in Yunnan's Minority Areas to Fulfill Social Responsibility
International Conference on Informatization in Education, Management and Business (IEMB 2015) The Co-construction of Mine Villages—an Important Way for Coal Mining Enterprises in Yunnan's Minority Areas to Fulfill Social Responsibility Yunhong GONG Qujing Normal University, Qujing, Yunnan Province, 655011 China Abstract. In the minority areas with the enterprises possess a capital advantage and the backward development of economy, the management personnel who are good at fulfillment of social responsibility by coal mining economic situation, planning, and innovation. enterprises is of more important significance. Therefore, the coal mining enterprises must be The coal mining enterprises in minority areas encouraged and supported to fulfill their social should pay more attention to the social responsibility and obligations, and give full play responsibility with the local minority to the advantages in capital, awareness in talents characteristics, mainly including maintaining the and development, and management ideas, etc. national unity, promoting the prosperity and Specifically, these enterprises can help development, and protecting the ecological collaborative villages know well the environment in minority areas, etc. In recent development ideas, make work measures, make years, explorations on a model for the every effort to the co-development of social co-construction of mine villages in many places undertakings and the co-construction of of Yunnan province are worthy of reference and infrastructure, actively undertake poverty introduction. Therefore, the co-construction of alleviation work, and participate in social mine villages is necessarily used as an important construction and management by combining with way for the coal mining enterprises in Yunnan's their own development characteristics and the minority areas to fulfill social responsibility. -

Household Stove Improvement and Risk of Lung Cancer in Xuanwei, China
Household Stove Improvement and Risk of Lung Cancer in Xuanwei, China Qing Lan, Robert S. Chapman, Dina M. Schreinemachers, Linwei Tian, Xingzhou He erably lower in China (6.8 in men and 3.2 in women) and was Background: Lung cancer rates in rural Xuanwei County, lower still in Yunnan as a whole (4.3 in men and 1.5 in women). Yunnan Province, are among the highest in China. Residents In Xuanwei, more than 90% of residents are farmers with little traditionally burned “smoky” coal in unvented indoor or no exposure to industrial or automotive air pollution, and firepits that generated very high levels of air pollution. Since residential stability is very high. Most men, but very few the 1970s, most residents have changed from firepits to women, smoke tobacco. Nearly all women, and some men, cook stoves with chimneys. This study assessed whether lung can- food on the household stove. cer incidence decreased after this stove improvement. Meth- For household cooking and heating, Xuanwei residents have ods: A cohort of 21 232 farmers, born from 1917 through traditionally burned “smoky coal,” “smokeless coal,” or wood in 1951, was followed retrospectively from 1976 through 1992. unvented indoor firepits. (Smoky coal and smokeless coal are All subjects were users of smoky coal who had been born general descriptive terms used throughout China for bituminous into homes with unvented firepits. During their lifetime, and anthracite coal, respectively.) Burning smoky coal in a 17 184 subjects (80.9%) changed permanently to stoves with firepit generates very high indoor concentrations of airborne 3 chimneys. -

Epidermal Growth Factor Receptor (EGFR) Mutations in Non-Small Cell Lung Cancer (NSCLC) of Yunnan in Southwestern China
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 9), pp: 15023-15033 Research Paper Epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer (NSCLC) of Yunnan in southwestern China Yongchun Zhou1,*, Yanlong Yang2,*, Chenggang Yang3,*, Yunlan Chen4, Changshao Yang5, Yaxi Du5, Guangqiang Zhao2,Yinjin Guo5, Lianhua Ye2, Yunchao Huang1,2 1Tumor Research Institute of Yunnan Province, The Third Affiliated Hospital of Kunming Medical University (Yunnan Tumor Hospital), Kunming, 650118, PR China 2Department of Thoracic Surgery I, The Third Affiliated Hospital of Kunming Medical University (Yunnan Tumor Hospital), Kunming, 650118, PR China 3Department of Pathology, The Third Affiliated Hospital of Kunming Medical University (Yunnan Tumor Hospital), Kunming, 650118, PR China 4Cadre Ward, The Third Affiliated Hospital of Kunming Medical University (Yunnan Tumor Hospital), Kunming, 650118, PR China 5Key Laboratory of Lung Cancer Research of Yunnan Province, The Third Affiliated Hospital of Kunming Medical University, Kunming, 650118, PR China *These authors contributed equally to this work Correspondence to: Yunchao Huang, email: [email protected] Keywords: non-small cell lung cancer, EGFR mutation, cftDNA, Yunnan, Xuanwei Received: September 22, 2016 Accepted: November 18, 2016 Published: January 17, 2017 ABSTRACT To investigate the Epidermal Growth Factor Receptor (EGFR) mutation status in non-small cell lung cancer (NSCLC) in Yunnan province in southwestern China, we detected EGFR mutation by Amplification Refractory Mutation System (ARMS) polymerase chain reaction (PCR) using DNA samples from 447 pathologically confirmed NSCLC specimens (175 tissue, 256 plasma and 16 cytologic samples). The relationship between EGFR mutations and demographic and clinical factors were further explored. Subgroup analyses according to sample type (tissue and plasma) and histological type (adenocarcinoma) were done. -

Yunnan Provincial Highway Bureau
IPP740 REV World Bank-financed Yunnan Highway Assets management Project Public Disclosure Authorized Ethnic Minority Development Plan of the Yunnan Highway Assets Management Project Public Disclosure Authorized Public Disclosure Authorized Yunnan Provincial Highway Bureau July 2014 Public Disclosure Authorized EMDP of the Yunnan Highway Assets management Project Summary of the EMDP A. Introduction 1. According to the Feasibility Study Report and RF, the Project involves neither land acquisition nor house demolition, and involves temporary land occupation only. This report aims to strengthen the development of ethnic minorities in the project area, and includes mitigation and benefit enhancing measures, and funding sources. The project area involves a number of ethnic minorities, including Yi, Hani and Lisu. B. Socioeconomic profile of ethnic minorities 2. Poverty and income: The Project involves 16 cities/prefectures in Yunnan Province. In 2013, there were 6.61 million poor population in Yunnan Province, which accounting for 17.54% of total population. In 2013, the per capita net income of rural residents in Yunnan Province was 6,141 yuan. 3. Gender Heads of households are usually men, reflecting the superior status of men. Both men and women do farm work, where men usually do more physically demanding farm work, such as fertilization, cultivation, pesticide application, watering, harvesting and transport, while women usually do housework or less physically demanding farm work, such as washing clothes, cooking, taking care of old people and children, feeding livestock, and field management. In Lijiang and Dali, Bai and Naxi women also do physically demanding labor, which is related to ethnic customs. Means of production are usually purchased by men, while daily necessities usually by women. -

Yunnan WLAN Hotspots 1/15
Yunnan WLAN hotspots NO. SSID Location_Name Location_Type Location_Address City Province 1 ChinaNet CuiHu and the surrounding area on foot Others CuiHu and the surrounding area on foot Kunming Yunnan 2 ChinaNet Hongta Sports Training Base Others Hongta Sports Training Base Kunming Yunnan 3 ChinaNet Center for Business Office Others No. 439 Beijing Road Kunming Kunming Yunnan 4 ChinaNet TaiLi business hall Others No. 39 South ring Road, Kunming City Kunming Yunnan 5 ChinaNet However, even the tranquility Board business hall Others However, even the town of Anning City even Ran Street No. 201 Kunming Yunnan 6 ChinaNet Dongchuan Village Road business hall Others Dongchuan Village Road, on the 17th Kunming Yunnan 7 ChinaNet Kunyang business hall Others Jinning County Kunyang the middle of the street Kunming Yunnan 8 ChinaNet Closing the business hall Others South Guandu District of Kunming customs in the next one (no No.) Kunming Yunnan 9 ChinaNet Songming county hall Others Songming County Huanglongbing Street I Kunming Yunnan 10 ChinaNet XUNDIAN Board Office of new business Others The new county transit roadside Telecom Tower, 1st Floor, (no number) Kunming Yunnan 11 ChinaNet New Asia Sports City stadium area Press Release Exhibition&stadium center Kunming Kwong Fuk Road and KunRei Road Kunming Yunnan 12 ChinaNet Kunming train the new South Station Hou car Room Railway Station/Bus Station Beijing Road South kiln Kunming Yunnan 13 ChinaNet Kunming Airport Airport KunMing Wujiaba Kunming Yunnan 14 ChinaNet Huazhou Hotel Hotel 223 East Road, Kunming City Kunming Yunnan 15 ChinaNet Kam Hotel Hotel 118 South Huan Cheng Road Kunming Kunming Yunnan 16 ChinaNet Greek Bridge Hotel Hotel Kunming Jiangbin West Road on the 1st Kunming Yunnan 17 ChinaNet Tyrone Hong Rui Hotel Hotel Kunming Spring City Road, No. -

Density Structure of the Crust in the Emeishan Large Igneous Province Revealed by the Lijiang- Guiyang Gravity Profile
Earth and Planetary Physics RESEARCH ARTICLE 2: 74–81, 2018 SOLID EARTH: GEODESY AND GRAVITY doi: 10.26464/epp2018007 Density structure of the crust in the Emeishan large igneous province revealed by the Lijiang- Guiyang gravity profile Xi Zhang1*, Peng Wang2, Tao Xu1,3, Yun Chen1, José Badal4, and JiWen Teng1 1State Key Laboratory of Lithospheric Evolution, Institute of Geology and Geophysics, Chinese Academy of Sciences, Beijing 100029, China; 2Institute of Earthquake Science, China Earthquake Administration, Beijing 100036, China; 3CAS Center for Excellence in Tibetan Plateau Earth Sciences, Beijing 100101, China; 4Physics of the Earth, Sciences B, University of Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain Abstract: The Emeishan large igneous province (hereafter named by its acronym ELIP) is the first accepted large igneous region in China. The current study tries to reconstruct the density structure of the crust in this region. For this purpose, we conducted the gravity survey along an 800-km-long profile, which stretched laterally along the latitude 27°N from Lijiang (Yunnan province) to Guiyang (Guizhou province). The fieldwork included 338 gravity measurements distributed from the inner zone to the outer zone of the mantle plume head. After a series of gravity reductions, we calculated the Bouguer gravity anomaly and then constructed the density model for ELIP by iterative forward modeling from an initial density model tightly constrained by wide-angle seismic reflection data. The topography of the Moho, here physically interpreted as a density discontinuity of ~0.4 g·cm–3, gradually rises from the inner zone (~50 km deep) to the outer zone (~40 km), describes a thicker crust in the inner zone than in any other segment of the profile and largely reproduces the shape of the Bouguer gravity anomaly curve. -

Agricultural, Ecological, and Social Insights: Residual Mulch Film Management Capacity and Policy Recommendations Based on Evidence in Yunnan Province, China
sustainability Article Agricultural, Ecological, and Social Insights: Residual Mulch Film Management Capacity and Policy Recommendations Based on Evidence in Yunnan Province, China Ji Chen 1,†, Xiao Chen 2,3,†, Jin Guo 4, Runyun Zhu 5, Mengran Liu 4, Xixi Kuang 6,7, Wenqing He 8,* and Yao Lu 2,* 1 College of Economic and Management, Yunnan Agricultural University, Kunming 650201, China; [email protected] 2 Institute of New Rural Development, Yunnan Agricultural University, Kunming 650201, China; [email protected] 3 State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Kunming 650201, China 4 Agricultural Environmental Protection and Monitoring Station of Yunnan Province, Kunming 650201, China; [email protected] (J.G.); [email protected] (M.L.) 5 Agricultural Technology Extension Station of Yunnan Province, Kunming 650201, China; [email protected] 6 Sichuan Ecology and Environment Cooperation Center, Chengdu 610000, China; [email protected] 7 Sichuan Ecology and Environment Protection Investment Evaluation and Performance Evaluation Center, Chengdu 610000, China 8 Key Laboratory for Prevention and Control of Residual Pollution in Agricultural Film, Ministry of Agriculture and Rural Affairs, Beijing 100081, China * Correspondence: [email protected] (W.H.); [email protected] (Y.L.) † These authors contributed equally to this work, and Xiao Chen shares first authorship. Abstract: Mulch film contaminates the environment while increasing agricultural yield. As such, the factors that impact the residual film management capacity of farmers must be identified, which would also be helpful for the sustainable development and security of agriculture. We in- Citation: Chen, J.; Chen, X.; Guo, J.; Zhu, R.; Liu, M.; Kuang, X.; He, W.; vestigated 10 counties across nine cities (states) by surveying 1284 households. -

WSB2 As a Target of Hedgehog Signaling Promoted the Malignant Biological Behavior of Xuanwei Lung Cancer Through Regulating Wnt/Β-Catenin Signaling
7404 Original Article WSB2 as a target of Hedgehog signaling promoted the malignant biological behavior of Xuanwei lung cancer through regulating Wnt/β-catenin signaling Xueqiang Wei1,2, Jun Liao1,2, Yujie Lei2, Minjie Li1,2, Guangqiang Zhao2, Yongchun Zhou2, Lianhua Ye2, Yunchao Huang2 1Kunming Medical University, Kunming, China; 2Department of Thoracic Surgery I, The Third Affiliated Hospital of Kunming Medical University/ Yunnan Cancer Hospital, Yunnan Cancer Center, The International Cooperation Key Laboratory of Regional Tumor in High Altitude Area, Kunming, China Contributions: (I) Conception and design: X Wei; (II) Administrative support: Y Huang; (III) Provision of study materials or patients: X Wei, Y Lei, G Zhao, L Ye; (IV) Collection and assembly of data: X Wei, M Li; (V) Data analysis and interpretation: All authors; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Yunchao Huang. Department of Thoracic Surgery I, The Third Affiliated Hospital of Kunming Medical University/Yunnan Cancer Hospital, No. 519, Kunzhou Road, Kunming 650118, China. Email: [email protected]. Background: Lung cancer represents the most leading causes of cancer-related deaths worldwide, especially in Xuanwei in eastern Yunnan province, China. WD repeat and SOCS box containing protein (WSB) has been reported to participate in the carcinogenesis of lung cancer. However, there is no report about the role of WSB2 in the carcinogenesis and development of lung cancer in Xuanwei. Here, we investigated the functional role of WSB2 in Xuanwei lung cancer and uncovered its underlying molecular mechanisms. Methods: The expression of WSB2 in lung cancer cell lines and tissues were measured using quantitative reverse transcription-polymerase chain reaction (qRT-PCR). -

If You Have Time for but One Province in China, Yúnn
© Lonely Planet Publications 216 云南 YÚNNÁN YÚNNÁN Y ú n n á n We’ve said it before and we’ll say it again: if you have time for but one province in China, Yúnnán should be it. Strong words but hyperbole is remarkably understated when describ- ing Yúnnán. No other province can rival Yúnnán’s diversity in land and people. Guìzhoū is also an ethnic mosaic, Sìchuān’s rivers garner much of the Southwest’s glory and Guǎngxī’s scenery leaps from every encyclopaedia’s entry on China. Yet Yúnnán can top ’em all. Just gaze at a map. Yúnnán’s majestic, and often sacred, peaks thrust from the Tibetan ranges to the north, lush jungle lies a two-day bus ride south and a fertile plain spreads through the rest of the province. It’s also home to China’s highest number of species of flora and fauna – including 2500 varieties of wild flowers and plants – and known for its mild climate year-round. Indeed, the province’s nicknames include ‘Kingdom of Plants’ and ‘Garden of Heavenly Marvellous Flowers’; the capital’s nickname is ‘Spring City’. A huge attraction is the province’s astonishingly diverse populace. Home to nearly half of all China’s ethnic minorities, nearly 50% of the province is non-Han (Han are China’s main ethnic group). Village-hop this breathtaking province and greet a new minority group each day, many in time-capsule towns that you’ll never forget. Smacks of PR pulp? Well, just be prepared that if you start here, you may never get to another province. -

Leiming Li Partner
Leiming Li Partner Practice Areas: Dispute Resolution, Private Equity/Venture Capital, Mergers & Acquisitions E-mail: [email protected] Tel: 010-56500995 E-mail: Professional Experience Tel: Leiming Li is a partner at Merits & Tree. Prior to joining Merits & Tree, Mr. Li was a partner at Hawkhigh Law Firm. Mr. Li is well-versed in areas such as dispute resolution, M&A, corporations; real estate, etc. Mr. Li has handled a wide range of litigation business and has accumulated rich practical experience in various commercial disputes. In recent years, he has paid more attention to investment and financing M&A and disputes, corporate governance, financial risk project disposal, and so on. Many of these cases are groundbreaking new types of cases. Mr. Li is good at mastering the overall working thought and proceeding from the perspective of customer’s core concern, and always provides the legal service which meets the needs for the customer. Mr. Li also serves as the general legal adviser to large enterprises, state-owned enterprises and scientific research institutes, covering aviation, real estate, tourism, high-end equipment manufacturing and other fields. Education LL.B., China University of Political Science and Law Qualifications Mr. Li has been admitted to practice in the People’s Republic of China Working language Chinese and Russian Representative Cases 1. M&A, Reorganization, Restructuring - Restructuring project of Zhuhai Securities Co., Ltd. - Restructuring project of Zhuhai Commercial Bank Co., Ltd. - Overall restructuring of China Electronic Engineering Design Institute - Equity and project transfer of Beijing Qianglong Real Estate Development Co., Ltd. www.meritsandtree.com - Ownership structure adjustment of the subsidiaries of Beyondwinet Information Technology Co., Ltd. -
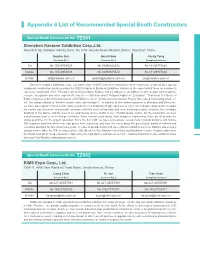
Appendix 6 List of Recommended Special Booth Constructors
Appendix 6 List of Recommended Special Booth Constructors Special Booth Constructor No. TZ001 Shenzhen Kastone Exhibition Corp.,Ltd. Room525, 5E, Software Industry Base, No.1004, Keyuan Road, Nanshan District, Shenzhen, China. Sandra Sun Gorsh Gao Cecily Yang Contacts No.1 Contacts No.2 Compliant Contact Tel 86-13530951029 86-18098925672 86-18129907680 Mobile 86-13530951029 86-18098925672 86-18129907680 E-Mail [email protected] [email protected] [email protected] Shenzhen Kastone Exhibition Corp., Ltd (stock code: 832971) has been honored for three consecutive years as No.1 special equipment construction service provider for CIIE Enterprise & Business Exhibition. Kastone is the expert which focus on customer’s experience and actual effect. Through years of accumulation, Kastone has a leading core strength in creative design, control system, resource integration and other aspects.We has the certification about “National High-tech Enterprise” “First-class Certificate of Engineering issued by China Association of Exhibition Centers” “Architectural Decoration Project Specialized Contracting Grade 2”, ect. Our design attitude is “efficient, simple, wise, and intelligent” . In addition to the creative agencies in Shanghai and Shenzhen, we have also signed contracts with many well-known multinational design agencies to carry out in-depth cooperation, integrate the world's top resources, and provide customers with the most cutting-edge and most matching creative solutions. Our company distribute in five places, and the area of our engineering service center is over 115,000 square meters. On the meanwhile, we have a professional team to serve foreign exhibitors. Since several years layout, from design to engineering, these are all provide the strong guarantee for the project operation. -

Administrative Division of Yunnan
Administrative Division of Yunnan Prefecture- County-level level Name Chinese (S) Hanyu Pinyin Panlong District ፧᰼ Pánlóng Qū Wuhua District ࡋ Wǔhuá Qū Guandu District Guāndù Qū Xishan District Xīshān Qū Dongchuan District Dōngchuān Qū Anning City ఓ Ānníng Shì Chenggong County Chénggòng Xiàn Kunming City ༷ఓ Jinning County Jìnníng Xiàn Kunming Shi Fumin County Fùmín Xiàn Yiliang County Yíliáng Xiàn Songming County Sōngmíng Xiàn Shilin Yi Autonomous ᕧ Shílín Yízú Zìzhìxiàn County Luquan Yi and Miao ᖾᕧ Lùquàn Yízú Autonomous County Miáozú Zìzhìxiàn Xundian Hui and Yi "#$ Xúndiàn Huízú Autonomous County Yízú Zìzhìxiàn ᕧ Qilin District ᯑ& Qílín Qū Qilin District ᯑ& Qílín Qū Xuanwei City '(ఓ Xuānwēi Shì Malong County Mǎlóng Xiàn Qujing City )᰼ ྍఓ Zhanyi County *፟ Zhānyì Xiàn Qǔjìng Shì Fuyuan County , Fùyuán Xiàn Luoping County -ఞ Luópíng Xiàn Shizong County ఙ0 Shīzōng Xiàn Luliang County 1 Lùliáng Xiàn Huize County 23 Huìzé Xiàn 2 Prefecture- County-level level Name Chinese (S) Hanyu Pinyin Hongta District ᐋ5 Hóngtǎ Qū Jiangchuan County 6 Jiāngchuān Xiàn Chengjiang County ၵ6 Chéngjiāng Xiàn Tonghai County 8ྦ Tōnghǎi Xiàn Huaning County Huáníng Xiàn Yuxi City ሊါఓ Yimen County : Yìmén Xiàn Yùxī Shì Eshan Yi < ᕧ Éshān Yízú Zìzhìxiàn Autonomous County Xinping Yi and Dai =ఞ Xīnpíng Yízú Autonomous County Dǎizú Zìzhìxiàn जᕧ Yuanjiang Hani, Yi ?6ૅA Yuánjiāng Hānízú and Dai Yízú Autonomous County जᕧ Dǎizú Zìzhìxiàn Longyang District ᬃC Lóngyáng Qū Shidian County Shīdiàn Xiàn Baoshan City D# ఓ Tengchong County Eউ Téngchōng Xiàn Bǎoshān