Australian Public Assessment Report for Pegaspargase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BC Cancer Benefit Drug List September 2021
Page 1 of 65 BC Cancer Benefit Drug List September 2021 DEFINITIONS Class I Reimbursed for active cancer or approved treatment or approved indication only. Reimbursed for approved indications only. Completion of the BC Cancer Compassionate Access Program Application (formerly Undesignated Indication Form) is necessary to Restricted Funding (R) provide the appropriate clinical information for each patient. NOTES 1. BC Cancer will reimburse, to the Communities Oncology Network hospital pharmacy, the actual acquisition cost of a Benefit Drug, up to the maximum price as determined by BC Cancer, based on the current brand and contract price. Please contact the OSCAR Hotline at 1-888-355-0355 if more information is required. 2. Not Otherwise Specified (NOS) code only applicable to Class I drugs where indicated. 3. Intrahepatic use of chemotherapy drugs is not reimbursable unless specified. 4. For queries regarding other indications not specified, please contact the BC Cancer Compassionate Access Program Office at 604.877.6000 x 6277 or [email protected] DOSAGE TUMOUR PROTOCOL DRUG APPROVED INDICATIONS CLASS NOTES FORM SITE CODES Therapy for Metastatic Castration-Sensitive Prostate Cancer using abiraterone tablet Genitourinary UGUMCSPABI* R Abiraterone and Prednisone Palliative Therapy for Metastatic Castration Resistant Prostate Cancer abiraterone tablet Genitourinary UGUPABI R Using Abiraterone and prednisone acitretin capsule Lymphoma reversal of early dysplastic and neoplastic stem changes LYNOS I first-line treatment of epidermal -
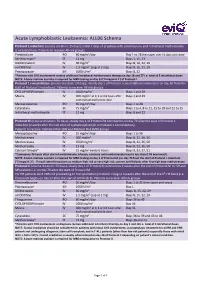
Acute Lymphoblastic Leukaemia: ALL06 Schema
Acute Lymphoblastic Leukaemia: ALL06 Schema Protocol 1 induction: (course duration: 35 days); initial 7 days of prephase with prednisolone and intrathecal methotrexate is included here. Patients to receive: All risk groups Prednisolone PO 60 mg/m2/day Day 1 to 28 then taper over 10 days and cease Methotrexate* IT 12 mg Days 1, 15, 33 DAUNOrubicin IV 30 mg/m2 Days 8, 16, 22, 29 vinCRISTine IV 1.5 mg/m2 (cap at 2 mg) Days 8, 16, 22, 29 Pegaspargase IM 1000 U/m2 Days 8, 22 *Patients with CNS involvement receive additional intrathecal methotrexate therapy on day 18 and 27 i.e. total of 5 intrathecal doses NOTE: A bone marrow aspirate is required for MRD testing on day 33 ('Timepoint 1') of Protocol I Protocol 1 consolidation: (course duration: 29 days); ideally day 1 of Protocol I consolidation commences on day 36 from the start of Protocol 1 induction ). Patients to receive: All risk groups. CYCLOPHOSPHamide IV 1000 mg/m2 Days 1 and 29 Mesna IV 400 mg/m2 at 0, 4 and 8 hours after Days 1 and 29 each cyclophosphamide dose Mercaptopurine PO 60 mg/m2/day Days 1 to 28 Cytarabine SC 75 mg/m2 Days 1 to 4, 8 to 11, 15 to 18 and 22 to 25 Intrathecal methotrexate IT 12 mg Days 8 and 22 Protocol M: (course duration: 56 days); ideally day 1 of Protocol M commences on day 79 after the start of Protocol 1 induction (2 weeks after the last dose of cyclophosphamide in Protocol 1 consolidation). -

Oncaspar, INN-Pegaspargase
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Oncaspar 750 U/ml solution for injection/infusion. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One ml of solution contains 750 Units (U)** of pegaspargase*. One vial of 5 ml solution contains 3,750 Units. * The active substance is a covalent conjugate of Escherichia coli-derived L-asparaginase with monomethoxypolyethylene glycol **One unit is defined as the quantity of enzyme required to liberate 1 µmol ammonia per minute at pH 7.3 and 37°C The potency of this medicinal product should not be compared to the one of another pegylated or non-pegylated protein of the same therapeutic class. For more information, see section 5.1. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Solution for injection/infusion. Clear, colourless solution. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Oncaspar is indicated as a component of antineoplastic combination therapy in acute lymphoblastic leukaemia (ALL) in paediatric patients from birth to 18 years, and adult patients. 4.2 Posology and method of administration Oncaspar should be prescribed and administered by physicians and/or health care personnel experienced in the use of antineoplastic products. It should only be given in a hospital setting where appropriate resuscitation equipment is available. Patients should be closely monitored for any adverse reactions throughout the administration period (see section 4.4). Posology Oncaspar is usually administered as part of combination chemotherapy protocols with other antineoplastic agents (see also section 4.5). Paediatric patients and adults ≤21 years The recommended dose in patients with a body surface area (BSA) ≥0.6 m2 and who are ≤21 years of age is 2,500 U of pegaspargase (equivalent to 3.3 ml Oncaspar)/m2 body surface area every 14 days. -

Prescribing Information | VELCADE® (Bortezomib)
HIGHLIGHTS OF PRESCRIBING INFORMATION Hypotension: Use caution when treating patients taking These highlights do not include all the information needed to antihypertensives, with a history of syncope, or with dehydration. use VELCADE safely and effectively. See full prescribing (5.2) information for VELCADE. Cardiac Toxicity: Worsening of and development of cardiac VELCADE® (bortezomib) for injection, for subcutaneous or failure has occurred. Closely monitor patients with existing heart intravenous use disease or risk factors for heart disease. (5.3) Initial U.S. Approval: 2003 Pulmonary Toxicity: Acute respiratory syndromes have ------------------------RECENT MAJOR CHANGES-------------------------- occurred. Monitor closely for new or worsening symptoms and consider interrupting VELCADE therapy. (5.4) Warnings and Precautions, Posterior Reversible Encephalopathy Syndrome: Consider MRI Thrombotic Microangiopathy (5.10) 04/2019 imaging for onset of visual or neurological symptoms; ------------------------INDICATIONS AND USAGE-------------------------- discontinue VELCADE if suspected. (5.5) VELCADE is a proteasome inhibitor indicated for: Gastrointestinal Toxicity: Nausea, diarrhea, constipation, and treatment of adult patients with multiple myeloma (1.1) vomiting may require use of antiemetic and antidiarrheal medications or fluid replacement. (5.6) treatment of adult patients with mantle cell lymphoma (1.2) Thrombocytopenia and Neutropenia: Monitor complete blood ----------------------DOSAGE AND ADMINISTRATION--------------------- -
Acute Lymphoblastic Leukemia (ALL) (Part 1 Of
LEUKEMIA TREATMENT REGIMENS: Acute Lymphoblastic Leukemia (ALL) (Part 1 of 12) Note: The National Comprehensive Cancer Network (NCCN) Guidelines® for Acute Lymphoblastic Leukemia (ALL) should be consulted for the management of patients with lymphoblastic lymphoma. Clinical Trials: The NCCN recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced healthcare team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are only provided to supplement the latest treatment strategies. The NCCN Guidelines are a work in progress that may be refined as often as new significant data becomes available. They are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any -

Negative Change Add Inclusion Criteria
Policy Drug(s) Type of Change Brief Description of Policy Change new policy Elitek (rasburicase) n/a n/a new policy Retevmo (selpercatinib) n/a n/a new policy Tabrecta (capmatinib) n/a n/a new policy Trodelvy (govitecan-hziy) n/a n/a new policy Qinlock (ripretinib) n/a n/a UM ONC_1048 Campath (alemtuzumab) Archive Campath is no longer being used for any Hematology Oncology indications. Remove inclusion criteria: ALL: Induction, consolidation, maitenance, relapsed/refractory combination removed and use is supported as a part of a multi-agent chemotherapy regimen, for all sub-types of ALL, for induction/consolidation therapy, and for therapy of relapsed refractory disease NHL: Induction, consolidation, maitenance, relapsed/refractory combination removed and use is supported for extra- nodal NK/T-cell lymphoma (nasal type) and as a part of a multi-agent chemotherapy regimen for either first line therapy and/or therapy for relapsed /refractory disease. UM ONC_1063 Oncaspar (pegaspargase) Positive change Remove exclusion criteria: History of serious thrombosis, pancreatitis, or hemorrhagic events with L-asparaginase (ELSPAR) therapy. UM ONC_1064 Oncaspar (pegaspargase) Positive change Add exclusion criteria: Dosing exceeds single dose limit of Trastuzumab 8 mg/kg for the loading dose, 6 mg/kg for UM ONC_1134 Trastuzumab products Negative change subsequent doses when given every 3 weeks; 4 mg/kg for the loading dose and 2 mg/kg for the weekly dose. Remove exclusion criteria: Velcade (bortezomib) is being used after disease progression on another Velcade-based regimen. UM ONC_1136 Velcade (bortezomib) Positive change Add inclusion criteria: Multiple Myeloma: - NOTE: The preferred agent, per NCH policy, is generic bortezomib over brand bortezomib (Velcade) for all indications, unless there are contraindications or intolerance to generic bortezomib. -

Standard Oncology Criteria C16154-A
Prior Authorization Criteria Standard Oncology Criteria Policy Number: C16154-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DATE DUE BEFORE 03/2016 12/2/2020 1/26/2022 HCPCS CODING TYPE OF CRITERIA LAST P&T APPROVAL/VERSION N/A RxPA Q1 2021 20210127C16154-A PRODUCTS AFFECTED: See dosage forms DRUG CLASS: Antineoplastic ROUTE OF ADMINISTRATION: Variable per drug PLACE OF SERVICE: Retail Pharmacy, Specialty Pharmacy, Buy and Bill- please refer to specialty pharmacy list by drug AVAILABLE DOSAGE FORMS: Abraxane (paclitaxel protein-bound) Cabometyx (cabozantinib) Erwinaze (asparaginase) Actimmune (interferon gamma-1b) Calquence (acalbrutinib) Erwinia (chrysantemi) Adriamycin (doxorubicin) Campath (alemtuzumab) Ethyol (amifostine) Adrucil (fluorouracil) Camptosar (irinotecan) Etopophos (etoposide phosphate) Afinitor (everolimus) Caprelsa (vandetanib) Evomela (melphalan) Alecensa (alectinib) Casodex (bicalutamide) Fareston (toremifene) Alimta (pemetrexed disodium) Cerubidine (danorubicin) Farydak (panbinostat) Aliqopa (copanlisib) Clolar (clofarabine) Faslodex (fulvestrant) Alkeran (melphalan) Cometriq (cabozantinib) Femara (letrozole) Alunbrig (brigatinib) Copiktra (duvelisib) Firmagon (degarelix) Arimidex (anastrozole) Cosmegen (dactinomycin) Floxuridine Aromasin (exemestane) Cotellic (cobimetinib) Fludara (fludarbine) Arranon (nelarabine) Cyramza (ramucirumab) Folotyn (pralatrexate) Arzerra (ofatumumab) Cytosar-U (cytarabine) Fusilev (levoleucovorin) Asparlas (calaspargase pegol-mknl Cytoxan (cyclophosphamide) -

Cancer Drug Costs for a Month of Treatment at Initial Food and Drug
Cancer drug costs for a month of treatment at initial Food and Drug Administration approval Year of FDA Monthly Cost Monthly cost Generic name Brand name(s) approval (actual $'s) (2014 $'s) vinblastine Velban 1965 $78 $586 thioguanine, 6-TG Thioguanine Tabloid 1966 $17 $124 hydroxyurea Hydrea 1967 $14 $99 cytarabine Cytosar-U, Tarabine PFS 1969 $13 $84 procarbazine Matulane 1969 $2 $13 testolactone Teslac 1969 $179 $1,158 mitotane Lysodren 1970 $134 $816 plicamycin Mithracin 1970 $50 $305 mitomycin C Mutamycin 1974 $5 $23 dacarbazine DTIC-Dome 1975 $29 $128 lomustine CeeNU 1976 $10 $42 carmustine BiCNU, BCNU 1977 $33 $129 tamoxifen citrate Nolvadex 1977 $44 $170 cisplatin Platinol 1978 $125 $454 estramustine Emcyt 1981 $420 $1,094 streptozocin Zanosar 1982 $61 $150 etoposide, VP-16 Vepesid 1983 $181 $430 interferon alfa 2a Roferon A 1986 $742 $1,603 daunorubicin, daunomycin Cerubidine 1987 $533 $1,111 doxorubicin Adriamycin 1987 $521 $1,086 mitoxantrone Novantrone 1987 $477 $994 ifosfamide IFEX 1988 $1,667 $3,336 flutamide Eulexin 1989 $213 $406 altretamine Hexalen 1990 $341 $618 idarubicin Idamycin 1990 $227 $411 levamisole Ergamisol 1990 $105 $191 carboplatin Paraplatin 1991 $860 $1,495 fludarabine phosphate Fludara 1991 $662 $1,151 pamidronate Aredia 1991 $507 $881 pentostatin Nipent 1991 $1,767 $3,071 aldesleukin Proleukin 1992 $13,503 $22,784 melphalan Alkeran 1992 $35 $59 cladribine Leustatin, 2-CdA 1993 $764 $1,252 asparaginase Elspar 1994 $694 $1,109 paclitaxel Taxol 1994 $2,614 $4,176 pegaspargase Oncaspar 1994 $3,006 $4,802 -

Velcade® (Bortezomib)
Velcade® (bortezomib) (Intravenous/Subcutaneous) Document Number: IC-0137 Last Review Date: 06/02/2020 Date of Origin: 11/28/2011 Dates Reviewed: 12/2011, 03/2012, 06/2012, 09/2012, 12/2012, 03/2013, 06/2013, 09/2013, 12/2013, 03/2014, 06/2014, 09/2014, 12/2014, 03/2015, 05/2015, 08/2015, 11/2015, 02/2016, 05/2016, 08/2016, 11/2016, 02/2017, 05/2017, 08/2017, 11/2017, 02/2018, 05/2018, 09/2018, 12/2018, 03/2019, 06/2019, 09/2019, 12/2019, 03/2020, 06/2020 I. Length of Authorization 1,5,6,8,14,25,26,30,35-41 Coverage will be provided for 6 months and may be renewed unless otherwise specified. • Initial treatment for Multiple Myeloma: Coverage will be provided for a total of 9 cycles (42-days per cycle). • Maintenance therapy in Multiple Myeloma for transplant-ineligible patients: Coverage will be provided for a total of 5 cycles (35-days per cycle). • Maintenance therapy in Multiple Myeloma post-autologous stem cell transplant: Coverage may be renewed up to 2 years of total therapy. • Re-treatment of Multiple Myeloma, initial treatment of Mantle Cell Lymphoma, & Adult T- Cell Leukemia/Lymphoma: Coverage will be provided for a total of 8 cycles (21-days per cycle). • Systemic Light Chain Amyloidosis as a single agent or in combination with cyclophosphamide and/or dexamethasone: Coverage will be provided for a total of 8 cycles (35-days per cycle as a single agent; 21- or 28-days per cycle in combination with cyclophosphamide and/or dexamethasone). -

Overview of the Current Treatment Strategy in Extranodal NK/T-Cell Lymphoma: from Diagnosis to Recurrence
10 Review Article Page 1 of 10 Overview of the current treatment strategy in extranodal NK/T-cell lymphoma: from diagnosis to recurrence Sang Eun Yoon, Seok Jin Kim, Won Seog Kim Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea Contributions: (I) Conception and design: All authors; (II) Administrative support: All authors; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: SE Yoon; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Won Seog Kim, MD, PhD. Division of Hematology and Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81, Irwon-ro, Gangnam-Gu, Seoul 06351, Korea. Email: [email protected]. Abstract: Extranodal natural killer/T cell lymphoma (ENKTL) is an extraordinary subtype of non- Hodgkin’s lymphoma (NHL), mainly involves the nasal cavity and the upper airway, and is influenced by the Epstein-Barr virus (EBV). Compared to other subtypes of T-cell lymphoma, ENKTL cannot be effectively treated with commonly used anthracycline-based systemic chemotherapy. Thus, the recent treatment of ENKTL has undergone significant changes based as informed by various clinical trials and prospective studies. Concurrent, sequential, and sandwich chemoradiotherapy, which is based on the alteration of the order of radiation and chemotherapy, has emerged as vital treatment approach for localized ENTKL. After the emergence of anthracycline-based systemic chemotherapy, nonanthracycline-based chemotherapy combined with L-asparaginase, including SMILE (dexamethasone, methotrexate, ifosfamide, L-asparaginase, and etoposide), Asp-MTX-Dex (L-asparaginase, methotrexate and dexamethasone), and DDGP (dexamethasone, cisplatin, gemcitabine, and peg-asparaginase) has attracted attention in advanced and relapsed/refractory (R/R) ENKTL treatment. -

Evaluation of Immunologic Crossreaction of Antiasparaginase Antibodies in Acute Lymphoblastic Leukemia (ALL) and Lymphoma Patien
Leukemia (2003) 17, 1583–1588 & 2003 Nature Publishing Group All rights reserved 0887-6924/03 $25.00 www.nature.com/leu Evaluation of immunologic crossreaction of antiasparaginase antibodies in acute lymphoblastic leukemia (ALL) and lymphoma patients w B Wang1, MV Relling2,3, MC Storm2, MH Woo3, R Ribeiro1,4, C-H Pui1,4, and LJ Hak2 1Department of Hematology and Oncology, St Jude Children’s Research Hospital, Memphis, TN, USA; 2Department of Pharmacy and the Center for Pediatric Pharmacokinetics and Therapeutics, College of Pharmacy, University of Tennessee, Memphis, TN, USA; 3Department of Pharmaceutical Sciences, St Jude Children’s, Research Hospital, Memphis, TN, USA; and 4College of Medicine, University of Tennessee, Memphis, TN, USA To evaluate how well antibodies to one asparaginase preparation pegaspargase asparaginase is modified from E. coli asparaginase predict or correlate with antibodies to another preparation in with covalent linkage to polyethylene glycol (PEG). Since acute lymphoblastic leukemia (ALL) and lymphoma patients who did and did not have hypersensitivity reactions during che- asparaginase is a foreign protein, the development of antiaspar- motherapy. In all, 24 children with newly diagnosed ALL or aginase antibodies frequently occurs. Antiasparaginase antibodies lymphoma, who received Escherichia coli asparaginase 10 000 IU/ may cause a decreased asparaginase activity through several m2 IM thrice weekly for nine doses as part of multiagent induction mechanisms. Antiasparaginase antibodies have been associated and reinduction chemotherapy, and seven monthly doses during with increased drug clearance thus shortening its half-life,8,9 the first 7 months of continuation treatment, were studied. decreasing circulating enzyme activity,10,11 or lowering enzyme Plasma samples were collected at postinduction and at post- activity by deposition of antigen–antibody complex in the reinduction. -

Haematologica August 2021
ARTICLE Acute Lymphoblastic Leukemia Pediatric-inspired chemotherapy incorporating Ferrata Storti Foundation pegaspargase is safe and results in high rates of minimal residual disease negativity in adults up to the age of 60 years with Philadelphia chromosome-negative acute lymphoblastic leukemia Haematologica 2021 1,2 3 4 5 Mark B. Geyer, Ellen K. Ritchie, Arati V. Rao, Shreya Vemuri, Jessica Volume 106(8):2086-2094 Flynn, 6 Meier Hsu6, Sean M. Devlin, 6 Mikhail Roshal, 7 Qi Gao, 7 Madhulika Shukla, 1 Jose M. Salcedo, 1 Peter Maslak, 1 Martin S. Tallman, 1 Dan Douer 1 and Jae H. Park 1,2 1Leukemia Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY; 2Center for Cell Engineering, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY; 3Weill Cornell Medical College, Hematology and Medical Oncology, Joan and Sanford I. Weill Department of Medicine, New York, NY; 4Kite-A Gilead Company, Foster City, CA; 5Sloan Kettering Institute, New York, NY; 6Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, NY and 7Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, USA ABSTRACT dministration of pediatric-inspired chemotherapy to adults up to age 60 with acute lymphoblastic leukemia (ALL) is challenging in Apart due to toxicities of asparaginase as well as myelosuppression. We conducted a multi-center phase II clinical trial (clinicaltrials gov. Identifier: NCT01920737) investigating a pediatric-inspired regimen, based on the augmented arm of the Children’s Cancer Group 1882 proto - col, incorporating six doses of pegaspargase 2,000 IU/m 2, rationally syn - chronized to avoid overlapping toxicity with other agents.