Using Biomolecules to Separate Plutonium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

THE NATURAL RADIOACTIVITY of the BIOSPHERE (Prirodnaya Radioaktivnost' Iosfery)
XA04N2887 INIS-XA-N--259 L.A. Pertsov TRANSLATED FROM RUSSIAN Published for the U.S. Atomic Energy Commission and the National Science Foundation, Washington, D.C. by the Israel Program for Scientific Translations L. A. PERTSOV THE NATURAL RADIOACTIVITY OF THE BIOSPHERE (Prirodnaya Radioaktivnost' iosfery) Atomizdat NMoskva 1964 Translated from Russian Israel Program for Scientific Translations Jerusalem 1967 18 02 AEC-tr- 6714 Published Pursuant to an Agreement with THE U. S. ATOMIC ENERGY COMMISSION and THE NATIONAL SCIENCE FOUNDATION, WASHINGTON, D. C. Copyright (D 1967 Israel Program for scientific Translations Ltd. IPST Cat. No. 1802 Translated and Edited by IPST Staff Printed in Jerusalem by S. Monison Available from the U.S. DEPARTMENT OF COMMERCE Clearinghouse for Federal Scientific and Technical Information Springfield, Va. 22151 VI/ Table of Contents Introduction .1..................... Bibliography ...................................... 5 Chapter 1. GENESIS OF THE NATURAL RADIOACTIVITY OF THE BIOSPHERE ......................... 6 § Some historical problems...................... 6 § 2. Formation of natural radioactive isotopes of the earth ..... 7 §3. Radioactive isotope creation by cosmic radiation. ....... 11 §4. Distribution of radioactive isotopes in the earth ........ 12 § 5. The spread of radioactive isotopes over the earth's surface. ................................. 16 § 6. The cycle of natural radioactive isotopes in the biosphere. ................................ 18 Bibliography ................ .................. 22 Chapter 2. PHYSICAL AND BIOCHEMICAL PROPERTIES OF NATURAL RADIOACTIVE ISOTOPES. ........... 24 § 1. The contribution of individual radioactive isotopes to the total radioactivity of the biosphere. ............... 24 § 2. Properties of radioactive isotopes not belonging to radio- active families . ............ I............ 27 § 3. Properties of radioactive isotopes of the radioactive families. ................................ 38 § 4. Properties of radioactive isotopes of rare-earth elements . -

Properties of Selected Radioisotopes
CASE FILE COPY NASA SP-7031 Properties of Selected Radioisotopes A Bibliography PART I: UNCLASSIFIED LITERATURE NATIONAL AERONAUTICS AND SPACE ADMINISTRATION NASA SP-7031 PROPERTIES OF SELECTED RADIOISOTOPES A Bibliography Part I: Unclassified Literature A selection of annotated references to technical papers, journal articles, and books This bibliography was compiled and edited by DALE HARRIS and JOSEPH EPSTEIN Goddard Space Flight Center Greenbelt, Maryland Scientific and Technical Information Division / OFFICE OF TECHNOLOGY UTILIZATION 1968 USP. NATIONAL AERONAUTICS AND SPACE ADMINISTRATION Washington, D.C. PREFACE The increasing interest in the application of substantial quantities of radioisotopes for propulsion, energy conversion, and various other thermal concepts emphasizes a need for the most recent and most accurate information available describing the nuclear, chemical, and physical properties of these isotopes. A substantial amount of progress has been achieved in recent years in refining old and developing new techniques of measurement of the properties quoted, and isotope processing. This has resulted in a broad technological base from which both the material and information about the material is available. Un- fortunately, it has also resulted in a multiplicity of sources so that information and data are either untimely or present properties without adequately identifying the measurement techniques or describing the quality of material used. The purpose of this document is to make available, in a single reference, an annotated bibliography and sets of properties for nine of the more attractive isotopes available for use in power production. Part I contains all the unclassified information that was available in the literature surveyed. Part II is the classified counterpart to Part I. -

Measurement of Cerium in Human Breast Milk and Blood
Measurement of cerium in human breast milk and blood Vera Höllriegl, Montserrat Gonzáles-Estecha*, Elena M. Trasobares*, Augusto Giussani, Uwe Oeh, Miguel Angel Herraiz**, Bernhard Michalkea Institute of Radiation Protection and aInstitute of Ecological Chemistry, Helmholtz Zentrum München, German Research Center for Environmental Health, Ingolstädter Landstrasse 1, 85764 Neuherberg, Germany *Trace Element Unit, Department of Laboratory Medicine and **Department of Obstetrics and Gynaecology, Hospital Clinico Universitario San Carlos, 28040 Madrid, Spain Abstract The aim of this study was to evaluate the relationship between cerium content in human breast milk and blood plasma or serum and the influence due to the environmental exposure. Blood samples and breast milk at various stages of lactation, from 5 days to 51 weeks post partum, were donated by 42 healthy breast-feeding mothers from Munich, Germany and by 26 lactating Spanish mothers from Madrid at 4 weeks post partum. Inductively coupled plasma mass spectrometry was applied for the determination of cerium in the biological samples. Cerium concentration in the digested milk samples from Munich showed low values and the arithmetic mean values ranged between the quantification limit of 5 ng/L up to 65 ng/L. The median value amounted to 13 ng/L. The cerium concentrations in the Spanish breast milk samples amounted to similar low values. The data were about a factor of eight lower than values found in a former study of samples from an eastern German province. All cerium concentrations in the German plasma samples, except for two, were at the quantification limit of 10 ng/L. Interestingly, the serum samples of the Spanish mothers 1 showed cerium values ranging between 21.6 and 70.3 ng/L; these higher data could be explained by an enhanced intake of cerium by humans in Madrid. -
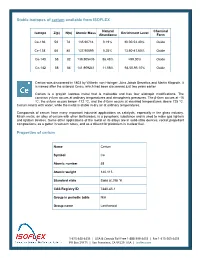
Stable Isotopes of Cerium Available from ISOFLEX Properties of Cerium
Stable isotopes of cerium available from ISOFLEX Natural Chemical Isotope Z(p) N(n) Atomic Mass Enrichment Level Abundance Form Ce-136 58 78 135.90714 0.19% 30.00-53.40% Oxide Ce-138 58 80 137.90599 0.25% 13.60-41.60% Oxide Ce-140 58 82 139.905435 88.48% >99.00% Oxide Ce-142 58 84 141.909241 11.08% 93.50-95.10% Oxide Cerium was discovered in 1803 by Wilhelm von Hisinger, Jöns Jakob Berzelius and Martin Klaproth. It is named after the asteroid Ceres, which had been discovered just two years earlier. Cerium is a grayish lustrous metal that is malleable and has four allotropic modifications. The common γ-form occurs at ordinary temperatures and atmospheric pressures. The β-form occurs at -16 ºC, the α-form occurs below -172 ºC, and the δ-form occurs at elevated temperatures above 725 ºC. Cerium reacts with water, while the metal is stable in dry air at ordinary temperatures. Compounds of cerium have many important industrial applications as catalysts, especially in the glass industry. Misch metal, an alloy of cerium with other lanthanides, is a pyrophoric substance and is used to make gas lighters and ignition devices. Some other applications of the metal or its alloys are in solid-state devices, rocket propellant compositions, as a getter in vacuum tubes, and as a diluent for plutonium in nuclear fuel. Properties of cerium Name Cerium Symbol Ce Atomic number 58 Atomic weight 140.115 Standard state Solid at 298 °K CAS Registry ID 7440-45-1 Group in periodic table N/A Group name Lanthanoid 1‐415‐440‐4433 │ USA & Canada Toll Free 1‐888‐399‐4433 -

19660018236.Pdf
NASA PRHS- pt .1 c.1 F1NA.L REPORT PROPERTIES OF RADIOISOTOPE HEAT SOURCES Contract NAS 5-9156 by COOK ELECTRIC COMPANY TECH-CENTER DIVISION MORTONGROVE, ILLINOIS Prepared for NATIONAL AERONAUTICS AND SPACE ADMINISTRATION , GODDARD SPACE FLIGHT CENTER , ,. ADVANCED POWER SOURCES SECTION GREENBELT, MARYLAND r -- ~ NASA CR-75439 ' Source: STAR, I v.4 81.5. TECH LIBRARY KAFB. NM FINAL REPORT PROPERTIES OF RADIOISOTOPE HEAT SOURCES 25 March 1965 to 31 August 1965 PART I (UNCLASSIFIED) Contract NAS 5-9156 COOK ELECTR.IC COMPANY TECH-CENTER DIVISION MORTONGROVE, ILLINOIS Pr epa red for NATIONAL AERONAUTICS AND SPACE ADMINISTRATION GODDARD SPACE FLIGHT CENTER ADVANCED POWER SOURCES SECTION GREENBELT, MAR-YLAND FINAL REPORT TITLE : Properties of Radioisotope Heat Sources CONTRACTOR Tech-CenterDivision Cook Electric Company Morton Grove, Illinois PERIOD: From 25 March 1965 to 31 August 1965 CLIENT:National Aeronautics and Space Administration Goddard Space Flight Center Advanced Power Sources Section Greenbelt,Maryland Mr. Dale Harris, Program Manager CONTRACT: NAS 5-9156 Program Manager D. E. Lehd6, Manager Undersea Warfare and Instrumentation Section ABSTRACT A report outlining the best available unclassified information on the nuclear, chemical, and physical properties of nine SNAP isotopes was prepared for NASA/GSFC Greenbelt, Maryland, Advanced Power Sourc.es Section. The isotopes reviewed are: Sr-90, Cs-134,Cs-137, Ce-144,Pm-147, Po-210, Pu-238, Cm-242, Cm-244. The properties reviewed were (1) Half Life; (2) Neutrons/Spontaneous Fission; (3) Neutrons from Spontaneous Fission; (4) Other sources of Radiation; (5) Energy Levels and Decay Schemes; (6) Fuel Forms; (7) Material Compatibility; (8) Effects of Impurities; (9) Thermal Conductivity; (10) Power Density; (11) Specific Power; (12) Heat Capacity; (13) Heat of Fusion; (14) Weight Density; (15) Melting Point; (16) Boiling Point; (17) Specific Activity; (18) Isotope Production, Availability,and Cost. -

Separation of Cerium from Lanthanum
Banha University Faculty of Science Chemistry Department Radiochemical Study on the Medically and Technologically Radionuclides of Some Lanthanides Presented by Hany Abd ElEl----HamidHamid Abd ElEl----AzizAziz Aglan Egyption Atomic Energy Authority Nuclear Research Center Cyclotron Project For The degree of Master of Science in Chemistry (Physical chemistry) Supervised by Prof. Dr. Mahmoud Ahmed Mousa Prof. Dr. Zeinab Abdou Saleh Professor of Physical Chemistry Professor of Nuclear Physics Faculty of Science Nuclear Research Center Benha University Atomic Energy Authority Dr. Hassan Ali Hanafi Lecturer of Physical and Applied Chemistry Cyclotron Project Nuclear Research Center Atomic Energy Authority 2010 Approval Sheet Title : Radiochemical Study on the Medically and Technologically Radionuclides of Some Lanthanides Name: Hany Abd ElEl----HamidHamid Abd ElEl----AzizAziz Aglan Supervisors: Name Position Signature Prof. Dr. Mahmoud Ahmed Professor of Physical Mousa Chemistry - Benha University Prof. Dr. Zeinab Abdou Professor of Nuclear Physics Saleh Atomic Energy Authority Dr. Hassan Ali Hanafi Lecturer of Physical and Applied Chemistry - Atomic Energy Authority Head of Chemistry Vice – Dean Dean of Faculty Department for Graduate Studies and Research Prof. Dr. S. G. Donia Prof. Dr. M. A. El-Fakharany REFEREES DECISION Title: Radiochemical Study on the Medically and Technologically Radionuclides of Some Lanthanides Name: Hany Abd El El----HamidHamid Abd ElEl----AzizAziz Aglan Referees: Name Position Signature Date of Discussion: Degree -

Production, Study and Us© of in Pure and Applied Nuclear Research
Production, study and us© of In pure and applied nuclear research by Tor Bjernstad University of Bergen 1986 THESIS for the degree Doctor Philosophiae (dr.philos.) at the University of Oslo. Obligatoric lectures: April 25, 1986. Defence of the thesis: April 26, 1986. to tlie thesis PRODUCTION, STUDI AND USE Of SHORT-LIVED NUCLIUES IH PURE AND APPLIED NUCLEAR RESEARCH by lor BJornstad LOCA!ION IS WRITTEN SHOULD BE 238,, • 235,, REVIEW PAPER, (>.)), line II "...Induced fission of U..." "...Induced fission of U..." REVIEW PAPER, p.14, Une 4 "Chemical metods..." "Chemical methods.,." SEVIEW PAPER, p.JO, line 17 "lived mud ides ("or..." "livpd nuclides for..." REVIEW PAPER, p.29, line 11 '...for sampling of this,.," "...for sampling of thin..." REVIEW PAPEfl, p.30, line 26 \..T1/2(M) * Tl/JID)..." \..Tj{H) * Tj(D)..." REVIEW PAPER, p.31, line 11 "...suncrocyclotron..," "... synchrocyclotron.*." REVIEW PAPER, p.31. line B **... Trie release tfie »uclear..." "...The release of the nuclear. from the bottom REVIEW PAPER, p.31. line A "...low vapor pressure..." ..low vapour pressure., from the bottom REVIEW PAPER, p.35, line •'...76Or(tj-1.355)..." \..76V(tj-1.3Ss)..." REVIEV PAPER, p.47, 1 lues 6, Ptiranthese after the reference on These parflntfieses should be on the 11,1? and 14 ttje level of Hie text line level of the reference number. In addition, there should be Inserted connid between references in line 15 and 12. REVIEW PAPER, p.48. line 1 "The separted molecular. "The separated molecular,.," PfVIEW PAPER, p.48, line 7 "...irridfum..." "...iridium..." from the bottom REVIEW PACER, p.50. -

1 Original Article 1 2 Vera Höllriegl, Astrid Barkleit, Vladimir Spielmann
1 Original Article 2 3 Vera Höllriegl, Astrid Barkleit, Vladimir Spielmann, Wei Bo Li 4 5 Measurement, model prediction and uncertainty quantification of plasma clearance of cerium citrate 6 in humans 7 8 Vera Höllriegl (),Vladimir Spielmann, Wei Bo Li 9 Institute of Radiation Medicine, Helmholtz Zentrum München, German Research Center for 10 Environmental Health, Ingolstädter Landstrasse 1, 85764 Neuherberg, Germany 11 E-mail: [email protected] 12 Phone: 0049-(0)89-3187-3219 13 Fax: 0049-(0)89-3187-3846 14 15 Astrid Barkleit 16 Institute of Resource Ecology, Helmholtz-Zentrum Dresden - Rossendorf, Bautzner Landstraße 400, 17 01328 Dresden, Germany 18 19 20 1 21 Abstract Double tracer studies in healthy human volunteers with stable isotopes of cerium citrate were 22 performed with the aim of investigating the gastro-intestinal absorption of cerium (Ce), its plasma 23 clearance and urinary excretion. In the present work, results of the clearance of Ce in blood plasma are 24 shown after simultaneous intravenous and oral administration of a Ce tracer. Inductively coupled 25 plasma mass spectrometry was used to determine the tracer concentrations in plasma. The results show 26 that about 80% of the injected Ce citrate cleared from the plasma within the five minutes post- 27 administration. The data obtained are compared to a revised biokinetic model of cerium, which was 28 initially developed by the International Commission on Radiological Protection (ICRP). The measured 29 plasma clearance of Ce citrate was mostly consistent with that predicted by the ICRP biokinetic model. 30 Furthermore, in an effort to quantify the uncertainty of the model prediction, the laboratory animal 31 data on which the ICRP biokinetic Ce model is based, was analyzed. -

University of California
UNIVERSITY OF CALIFORNIA tlo TWO-WEEK lOAN COPY This is a Library Circuldi:ing Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Dioision, Ext. 5545 BERKELEY. CALIFORNIA DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of aut~ors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. .). : . .. UNDnmSITY OF CALIFORNIA R1:,DIATION LABOR/iTORY Cover Sheet INDEX t·lO.l )(lt'\, L- l 0 9 Do not rew~ove This d,ocument contains~pages and~ol~tes of ~igures. This is copyf..Zof!JJ.. :eriesA._ Issued to: ~~.~Ly. <;LASSIF1'~,\T10"1 (' •,l'>!~":Lt~D IW A~THORlTl'_ Q::O .,. : 1 .· : · ·• r ENG[;>lEea Each person whoaM~l~.,;~f~~~~'tl'Mgn the. -

University of California Ernest 0. Radiation Lawrence Laboratory
Lawrence Berkeley National Laboratory Recent Work Title COLLECTIVE EXCITATION OF NEUTRON-DEFICIENT BARIUM, XENON, AND CERIUM ISOTOPES: CENTRIFUGAL STRETCHING TREATMENT OF NUCLEI Permalink https://escholarship.org/uc/item/8p2539fp Author Clarkson, Jack E. Publication Date 1965-05-01 eScholarship.org Powered by the California Digital Library University of California UCRL-16040 University of California Ernest 0. Lawrence Radiation Laboratory TWO-WEEK lOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Division, Ext. 5545 COLLECTIVE EXCITATION OF NEUTRON -DEFICIENT BARIUM, XENON, AND CERIUM ISOTOPES: CENTRIFUGAL STRETCHING TREATMENT OF NUCLEI Berkeley, California DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. -

The Photoneutron Cross Section in the Giant Resonance Region for Several 82 Neutron Isotones Ronald Gordon Johnson Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1970 The photoneutron cross section in the giant resonance region for several 82 neutron isotones Ronald Gordon Johnson Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Nuclear Commons Recommended Citation Johnson, Ronald Gordon, "The hotp oneutron cross section in the giant resonance region for several 82 neutron isotones " (1970). Retrospective Theses and Dissertations. 4847. https://lib.dr.iastate.edu/rtd/4847 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. 71^4,235 lif&HNSON, Ronald Gordon, 1941^ THE PHOTONEUTRON CROSS SECTION IN THE GIANT RESONANCE REGION FOR SEVERAL 82 NEUTRON ISOTONES. Iowa State University, Ph.D., 1970 Physics, nuclear University Microfilms, A XEROX Company, Ann Arbor, Michigan THE PHOTONEUTRON CROSS SECTION IN THE GIANT RESONANCE REGION FOR SEVERAL 82 NEUTRON ISOTONES by Ronald Gordon Johnson A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject; Nuclear Physics Approved: Signature was redacted for privacy. Signature was redacted for privacy. Department Signature was redacted for privacy. Iowa State University Of Science and Technoiogy Ames., Iowa 1970 TABLE OF CONTENTS Page i. INTRODUCTION 1 II. GENERAL EXPERIMENTAL CONSIDERATIONS 8 A. Summary of Experimental Procedure 8 B. -

Discovery of Cesium, Lanthanum, Praseodymium and Promethium Isotopes
Discovery of Cesium, Lanthanum, Praseodymium and Promethium Isotopes E. May, M. Thoennessen∗ National Superconducting Cyclotron Laboratory and Department of Physics and Astronomy, Michigan State University, East Lansing, MI 48824, USA Abstract Currently, forty-one cesium, thirty-five lanthanum, thirty-two praseodymium, and thirty-one promethium, isotopes have been observed and the discovery of these isotopes is discussed here. For each isotope a brief synopsis of the first refereed publication, including the production and identification method, is presented. arXiv:1109.1784v1 [nucl-ex] 8 Sep 2011 ∗Corresponding author. Email address: [email protected] (M. Thoennessen) Preprint submitted to Atomic Data and Nuclear Data Tables October 29, 2018 Contents 1. Introduction . 2 2. Discovery of 112−152Cs.................................................................................. 3 3. Discovery of 117−153La.................................................................................. 11 4. Discovery of 121−154Pr.................................................................................. 20 5. Discovery of 128−158Pm................................................................................. 27 6. Summary ............................................................................................. 35 References . 35 Explanation of Tables . 44 Table 1. Discovery of cesium, lanthanum, praseodymium and promethium isotopes. See page 44 for Explanation of Tables 45 1. Introduction The discovery of cesium, lanthanum, praseodymium