Derivation of Sarcomas from Mesenchymal Stem Cells Via Inactivation of the Wnt Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Wnt Signaling in Vertebrate Neural Development and Function
J Neuroimmune Pharmacol (2012) 7:774–787 DOI 10.1007/s11481-012-9404-x INVITED REVIEW Wnt Signaling in Vertebrate Neural Development and Function Kimberly A. Mulligan & Benjamin N. R. Cheyette Received: 18 June 2012 /Accepted: 10 September 2012 /Published online: 27 September 2012 # Springer Science+Business Media, LLC 2012 Abstract Members of the Wnt family of secreted signaling immature neurons migrate to populate different cortical proteins influence many aspects of neural development and layers, nuclei, and ganglia in the forebrain, midbrain, hind- function. Wnts are required from neural induction and axis brain, and spinal cord. Each mature neuron elaborates an formation to axon guidance and synapse development, and axon and numerous dendrites while forming hundreds to even help modulate synapse activity. Wnt proteins activate a thousands of synapses with other neurons, enabling electro- variety of downstream signaling pathways and can induce a chemical communication throughout the nervous system. similar variety of cellular responses, including gene tran- All these developmental processes must be coordinated to scription changes and cytoskeletal rearrangements. This re- ensure proper construction and function of the CNS, and view provides an introduction to Wnt signaling pathways this requires the coordinated developmental activity of a and discusses current research on their roles in vertebrate vast array of genes. neural development and function. Members of the Wnt family of secreted signaling proteins are implicated in every step of neural development men- Keywords Wnt signaling . Neural development . tioned above. Wnt proteins provide positional information CNS patterning . Axon guidance . Dendrite growth . within the embryo for anterior-posterior axis specification of Synapse formation . -

Evolutionarily Conserved Tbx5–Wnt2/2B Pathway Orchestrates Cardiopulmonary Development
Evolutionarily conserved Tbx5–Wnt2/2b pathway orchestrates cardiopulmonary development Jeffrey D. Steimlea,b,c, Scott A. Rankind,e,f,g, Christopher E. Slagleh,i,j,k, Jenna Bekenya,b,c, Ariel B. Rydeena,b,c, Sunny Sun-Kin Chanl,m, Junghun Kweona,b,c, Xinan H. Yanga,b,c, Kohta Ikegamia,b,c, Rangarajan D. Nadadura,b,c, Megan Rowtona,b,c, Andrew D. Hoffmanna,b,c, Sonja Lazarevica,b,c, William Thomasn,o, Erin A. T. Boyle Andersonp, Marko E. Horbn,o, Luis Luna-Zuritaq,r, Robert K. Hom, Michael Kybal,m, Bjarke Jensens, Aaron M. Zornd,e,f,g, Frank L. Conlonh,i,j,k, and Ivan P. Moskowitza,b,c,1 aDepartment of Pediatrics, University of Chicago, Chicago, IL 60637; bDepartment of Pathology, University of Chicago, Chicago, IL 60637; cDepartment of Human Genetics, University of Chicago, Chicago, IL 60637; dCenter for Stem Cell and Organoid Medicine, Cincinnati Children’s Research Foundation, Cincinnati, OH 45229; eDepartment of Pediatrics, College of Medicine, University of Cincinnati, Cincinnati, OH 45229; fDivision of Developmental Biology, Perinatal Institute, Cincinnati Children’s Research Foundation, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, OH 45229; gDepartment of Pediatrics, College of Medicine, University of Cincinnati, Cincinnati, OH 45229; hDepartment of Biology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; iDepartment of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; jIntegrative Program for Biological and Genome Sciences, University of North -

REVIEW ARTICLE the Genetics of Autism
REVIEW ARTICLE The Genetics of Autism Rebecca Muhle, BA*; Stephanie V. Trentacoste, BA*; and Isabelle Rapin, MD‡ ABSTRACT. Autism is a complex, behaviorally de- tribution of a few well characterized X-linked disorders, fined, static disorder of the immature brain that is of male-to-male transmission in a number of families rules great concern to the practicing pediatrician because of an out X-linkage as the prevailing mode of inheritance. The astonishing 556% reported increase in pediatric preva- recurrence rate in siblings of affected children is ϳ2% to lence between 1991 and 1997, to a prevalence higher than 8%, much higher than the prevalence rate in the general that of spina bifida, cancer, or Down syndrome. This population but much lower than in single-gene diseases. jump is probably attributable to heightened awareness Twin studies reported 60% concordance for classic au- and changing diagnostic criteria rather than to new en- tism in monozygotic (MZ) twins versus 0 in dizygotic vironmental influences. Autism is not a disease but a (DZ) twins, the higher MZ concordance attesting to ge- syndrome with multiple nongenetic and genetic causes. netic inheritance as the predominant causative agent. By autism (the autistic spectrum disorders [ASDs]), we Reevaluation for a broader autistic phenotype that in- mean the wide spectrum of developmental disorders cluded communication and social disorders increased characterized by impairments in 3 behavioral domains: 1) concordance remarkably from 60% to 92% in MZ twins social interaction; 2) language, communication, and and from 0% to 10% in DZ pairs. This suggests that imaginative play; and 3) range of interests and activities. -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners. -

WNT2 and WNT7B Cooperative Signaling in Lung Development
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 WNT2 and WNT7B Cooperative Signaling in Lung Development Mayumi Miller University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Developmental Biology Commons, and the Molecular Biology Commons Recommended Citation Miller, Mayumi, "WNT2 and WNT7B Cooperative Signaling in Lung Development" (2012). Publicly Accessible Penn Dissertations. 674. https://repository.upenn.edu/edissertations/674 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/674 For more information, please contact [email protected]. WNT2 and WNT7B Cooperative Signaling in Lung Development Abstract The development of a complex organ, such as the lung, relies upon precisely controlled temporal and spatial expression patterns of signaling pathways for proper specification and differentiation of the cell types required to build a lung. While progress has been made in dissecting the network of signaling pathways and the integration of their positive and negative feedback mechanisms, there is still much to discover. For example, the Wnt signaling pathway is required for lung specification and growth, but a combinatorial role for Wnt ligands has not been investigated. In this dissertation, I combine mouse genetic models and in vitro and ex vivo lung culture assays, to determine a cooperative role for Wnt2 and Wnt7b in the developing lung. This body of work reveals the requirement of cooperative signaling between Wnt2 and Wnt7b for smooth muscle development and proximal to distal patterning of the lung. Additional findings er veal a role for the Pdgf pathway and homeobox genes in potentiating this cooperation. -
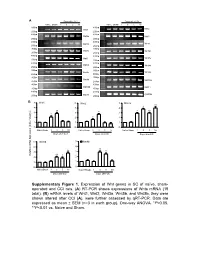
Supplementary Figure 1. Expression of Wnt Genes in SC of Naïve
A Days after CCI Days after CCI Naive Sham 1 3 5 10 Naive Sham 1 3 5 10 500bp 400bp Wnt1 Wnt2 250bp 250bp 400bp 400bp Wnt2b Wnt3 250bp 250bp 400bp 400bp Wnt3a Wnt4 250bp 250bp 300bp 400bp Wnt5a Wnt5b 200bp 250bp 400bp 400bp Wnt6 Wnt7a 250bp 250bp 400bp 400bp Wnt7b Wnt8a 250bp 250bp 400bp 350bp Wnt8b Wnt9a 250bp 200bp 400bp 400bp Wnt9b Wnt10a 250bp 250bp 400bp 400bp Wnt10b Wnt11 250bp 250bp 400bp 400bp Wnt16 GAPDH 250bp 250bp B 5 Wnt1 5 Wnt2 5 Wnt3a ** 4 4 4 * ** ** 3 3 * 3 * * 2 2 * 2 1 1 1 0 0 0 NaïveSham 1 3 5 10 Naïve Sham 1 3 5 10 Naïve Sham 1 3 5 10 Days after CCI Days after CCI Days after CCI 5 Wnt5b 5 Wnt8b 4 4 ** 3 ** 3 ** Relative mRNA Expression (fold of change) ** 2 * 2 1 1 0 0 NaïveSham 1 3 5 10 NaïveSham 1 3 5 10 Days after CCI Days after CCI Supplementary Figure 1 A B Negative Weak Moderate Strong Negative Weak Moderate Strong Large-sized cells 100% 100% 80% 80% 60% 60% 40% 20% 40% 0% 20% Sham CCI-1d CCI-14d Medium-sized cells Wnt3a immunoreactivity cells 0% 100% CGRP(+) IB4(+) 80% Small cells 60% 40% 20% 0% Sham CCI-1d CCI-14d Wnt3a immunoreactivity cells Small cells 100% 80% 60% 40% 20% 0% Sham CCI-1d CCI-14d Fig. S2 4 Fz1 4 Fz3 4 Fz4 4 Fz5 ** 3 3 ** 3 3 2 ** 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1 5 10 NaïveSham 1 5 10 Days after CCI Days after CCI Days after CCI Days after CCI 4 Fz6 4 Fz7 4 Fz8 4 Fz9 (Fold of Change) of (Fold 3 3 3 ** 3 Relative mRNA Expression Expression mRNA Relative ** 2 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1d 5d 10d NaïveSham 1 -

International Union of Basic and Clinical Pharmacology. LXXX. the Class Frizzled Receptors□S
Supplemental Material can be found at: /content/suppl/2010/11/30/62.4.632.DC1.html 0031-6997/10/6204-632–667$20.00 PHARMACOLOGICAL REVIEWS Vol. 62, No. 4 Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics 2931/3636111 Pharmacol Rev 62:632–667, 2010 Printed in U.S.A. International Union of Basic and Clinical Pharmacology. LXXX. The Class Frizzled Receptors□S Gunnar Schulte Section of Receptor Biology and Signaling, Department of Physiology and Pharmacology, Karolinska Institutet, Stockholm, Sweden Abstract............................................................................... 633 I. Introduction: the class Frizzled and recommended nomenclature............................ 633 II. Brief history of discovery and some phylogenetics ......................................... 634 III. Class Frizzled receptors ................................................................ 634 A. Frizzled and Smoothened structure ................................................... 635 1. The cysteine-rich domain ........................................................... 635 2. Transmembrane and intracellular domains........................................... 636 3. Motifs for post-translational modifications ........................................... 636 B. Class Frizzled receptors as PDZ ligands............................................... 637 C. Lipoglycoproteins as receptor ligands ................................................. 638 Downloaded from IV. Coreceptors........................................................................... -

Wnt8b Regulates Myo Broblast Differentiation of Lung-Resident Mesenchymal Stem Cells Via the Activation of Wnt/Β-Catenin Signal
Wnt8b Regulates Myobroblast Differentiation of Lung-Resident Mesenchymal Stem Cells via the Activation of Wnt/β-catenin Signaling in Pulmonary Fibrogenesis Chaowen Shi Nanjing University Medical School Xiang Chen Nanjing University Medical School Wenna Yin Nanjing University Medical School Jiwei Hou Nanjing University Medical School Zhaorui Sun Nanjing University Medical School Clinical College: East Region Military Command General Hospital Xiaodong Han ( [email protected] ) Nanjing University https://orcid.org/0000-0003-4512-3699 Research Keywords: Lung resident mesenchymal stem cell, Wnt8b, Wnt/β-catenin signaling, Idiopathic pulmonary brosis Posted Date: May 6th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-466908/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/19 Abstract Background Idiopathic pulmonary brosis (IPF) is a chronic, progressive, and fatal lung disease that is characterized by enhanced changes in stem cell differentiation and broblast proliferation. Lung resident mesenchymal stem cells (LR-MSCs) are important regulators of pathophysiological processes including tissue repair and inammation, and evidence suggests that this cell population also plays an essential role in brosis. Our previous study demonstrated that Wnt/β-catenin signaling is aberrantly activated in the lungs of bleomycin-treated mice and induces myobroblast differentiation of LR-MSCs. However, the underlying correlation between LR-MSCs and the Wnt/β-catenin signaling remains poorly understood. Methods We used mRNA microarray, immunohistochemistry assay, qRT-PCR, and western blotting to measure the expression of Wnt8b in myobroblast differentiation of LR-MSCs and BLM-induced mouse brotic lungs. Immunouorescence staining and western blotting were performed to analyze myobroblast differentiation of LR-MSCs after overexpressing or silence Wnt8b. -

Downregulation of Wnt2 and B-Catenin by Sirna
Cancer Gene Therapy (2009) 16, 351–361 r 2009 Nature Publishing Group All rights reserved 0929-1903/09 $32.00 www.nature.com/cgt ORIGINAL ARTICLE Downregulation of Wnt2 and b-catenin by siRNA suppresses malignant glioma cell growth PPu1, Z Zhang1, C Kang1, R Jiang1, Z Jia1, G Wang1 and H Jiang2 1Department of Neurosurgery, Tianjin Medical University General Hospital; Laboratory of Neuro-Oncology, Tianjin Neurological Institute, Tianjin, People’s Republic of China and 2Department of Neurology, Henry Ford Hospital, Detroit, MI, USA Increasing evidence suggests that aberrant activation of Wnt signaling is involved in tumor development and progression. Our earlier study on gene expression profile in human gliomas by microarray found that some members of Wnt family were overexpressed. To further investigate the involvement of Wnt signaling in gliomas, the expression of core components of Wnt signaling cascade in 45 astrocytic glioma specimens with different tumor grades was examined by reverse transcription-PCR and immunohistochemistry. Wnt2, Wnt5a, frizzled2 and b-catenin were overexpressed in gliomas. Knockdown of Wnt2 and its key mediator b-catenin in the canonical Wnt pathway by siRNA in human U251 glioma cells inhibited cell proliferation and invasive ability, and induced apoptotic cell death. Furthermore, treating the nude mice carrying established subcutaneous U251 gliomas with siRNA targeting Wnt2 and b-catenin intratumorally also delayed the tumor growth. In both in vitro and in vivo studies, downregulation of Wnt2 and b-catenin was associated with the decrease of PI3K/p-AKT expression, indicating the interplay between Wnt/b-catenin and PI3K/AKT signaling cascades. In conclusion, the canonical Wnt pathway is of critical importance in the gliomagenesis and intervention of this pathway may provide a new therapeutic approach for malignant gliomas. -

Wnt Ligands Are Not Required for Planar Cell Polarity in the Drosophila Wing Or Notum
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.05.137182; this version posted June 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Wnt ligands are not required for planar cell polarity in the Drosophila wing or notum Ben Ewen-Campen1, Typhaine Comyn1,2, Eric Vogt1, Norbert Perrimon1,3 Affiliations: 1. Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA 2. Present Address: Energy & Memory, Brain Plasticity Unit, Centre National de la Recherche Scientifique, ESPCI, Paris, France 3. Howard Hughes Medical Institute, Boston, MA 02115, USA Corresponding Author: Norbert Perrimon Harvard Medical School, New Research Bldg., Room 336G, Howard Hughes Medical Institute 77 Ave. Louis Pasteur, Boston, MA 02115. Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.05.137182; this version posted June 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The frizzled (fz) and disheveled (dsh) genes are highly conserved members of the core planar cell polarity (PCP) pathway and of the Wnt signaling pathway. Given these dual functions, a number of studies have examined whether Wnt ligands may provide a global, tissue-scale orientation cue for PCP establishment during development, and these studies have reached differing conclusions. In this study, we re-examine this issue in the Drosophila melanogaster wing and notum using split-Gal4 co-expression analysis, systematic pairwise and triple somatic CRISPR-based knock-outs and double RNAi experiments. -

Gene Amplifications at Chromosome 7 of the Human Gastric Cancer Genome
225-231 4/7/07 20:50 Page 225 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 20: 225-231, 2007 225 Gene amplifications at chromosome 7 of the human gastric cancer genome SANGHWA YANG Cancer Metastasis Research Center, Yonsei University College of Medicine, 134 Shinchon-Dong, Seoul 120-752, Korea Received April 20, 2007; Accepted May 7, 2007 Abstract. Genetic aberrations at chromosome 7 are known to Introduction be related with diverse human diseases, including cancer and autism. In a number of cancer research areas involving gastric The completion of human genome sequencing and the cancer, several comparative genomic hybridization studies subsequent gene annotations, together with a rapid develop- employing metaphase chromosome or BAC clone micro- ment of high throughput screening technologies, such as DNA arrays have repeatedly identified human chromosome 7 microarrays, have made it possible to perform genome-scale as containing ‘regions of changes’ related with cancer expression profiling and comparative genomic hybridizations progression. cDNA microarray-based comparative genomic (CGHs) in various cancer models. The elucidation of gene hybridization can be used to directly identify individual target copy number variations in several cancer genomes is generating genes undergoing copy number variations. Copy number very informative results. Metaphase chromosome CGH and change analysis for 17,000 genes on a microarray format was the recent introduction of BAC and especially cDNA micro- performed with tumor and normal gastric tissues from 30 array-based CGHs (aCGH) (1) have greatly contributed to patients. A group of 90 genes undergoing copy number the identification of chromosome aberrations and of amplified increases (gene amplification) at the p11~p22 or q21~q36 and deleted genes in gastric cancer tissues and cell lines (2-9). -

The Role of Wnt Signalling in Chronic Kidney Disease (CKD)
G C A T T A C G G C A T genes Review The Role of Wnt Signalling in Chronic Kidney Disease (CKD) Soniya A. Malik , Kavindiya Modarage and Paraskevi Goggolidou * Department of Biomedical Science and Physiology, Faculty of Science and Engineering, University of Wolverhampton, Wulfruna Street, Wolverhampton WV1 1LY, UK; [email protected] (S.A.M.); [email protected] (K.M.) * Correspondence: [email protected]; Tel.: +44-01902321152 Received: 9 March 2020; Accepted: 29 April 2020; Published: 30 April 2020 Abstract: Chronic kidney disease (CKD) encompasses a group of diverse diseases that are associated with accumulating kidney damage and a decline in glomerular filtration rate (GFR). These conditions can be of an acquired or genetic nature and, in many cases, interactions between genetics and the environment also play a role in disease manifestation and severity. In this review, we focus on genetically inherited chronic kidney diseases and dissect the links between canonical and non-canonical Wnt signalling, and this umbrella of conditions that result in kidney damage. Most of the current evidence on the role of Wnt signalling in CKD is gathered from studies in polycystic kidney disease (PKD) and nephronophthisis (NPHP) and reveals the involvement of β-catenin. Nevertheless, recent findings have also linked planar cell polarity (PCP) signalling to CKD, with further studies being required to fully understand the links and molecular mechanisms. Keywords: Wnt signalling; polycystic kidney disease; nephronophthisis; beta-catenin; planar cell polarity signalling 1. Chronic Kidney Disease (CKD) Chronic kidney disease (CKD) is a term that denotes the presence of kidney damage, presenting with abnormalities or a decline in kidney function.