Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Compositions and Methods for Selective Delivery of Oligonucleotide Molecules to Specific Neuron Types
(19) TZZ ¥Z_T (11) EP 2 380 595 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.10.2011 Bulletin 2011/43 A61K 47/48 (2006.01) C12N 15/11 (2006.01) A61P 25/00 (2006.01) A61K 49/00 (2006.01) (2006.01) (21) Application number: 10382087.4 A61K 51/00 (22) Date of filing: 19.04.2010 (84) Designated Contracting States: • Alvarado Urbina, Gabriel AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Nepean Ontario K2G 4Z1 (CA) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • Bortolozzi Biassoni, Analia Alejandra PT RO SE SI SK SM TR E-08036, Barcelona (ES) Designated Extension States: • Artigas Perez, Francesc AL BA ME RS E-08036, Barcelona (ES) • Vila Bover, Miquel (71) Applicant: Nlife Therapeutics S.L. 15006 La Coruna (ES) E-08035, Barcelona (ES) (72) Inventors: (74) Representative: ABG Patentes, S.L. • Montefeltro, Andrés Pablo Avenida de Burgos 16D E-08014, Barcelon (ES) Edificio Euromor 28036 Madrid (ES) (54) Compositions and methods for selective delivery of oligonucleotide molecules to specific neuron types (57) The invention provides a conjugate comprising nucleuc acid toi cell of interests and thus, for the treat- (i) a nucleic acid which is complementary to a target nu- ment of diseases which require a down-regulation of the cleic acid sequence and which expression prevents or protein encoded by the target nucleic acid as well as for reduces expression of the target nucleic acid and (ii) a the delivery of contrast agents to the cells for diagnostic selectivity agent which is capable of binding with high purposes. -

Certificate of Analysis
Print Date: Jan 8th 2016 Certificate of Analysis www.tocris.com Product Name: Guvacine hydrochloride Catalog No.: 0234 Batch No.: 13 CAS Number: 6027-91-4 IUPAC Name: 1,2,5,6-Tetrahydropyridine-3-carboxylic acid hydrochloride 1. PHYSICAL AND CHEMICAL PROPERTIES Batch Molecular Formula: C6H9NO2.HCl Batch Molecular Weight: 163.6 Physical Appearance: White crystalline solid Solubility: water to 100 mM phosphate buffered saline to 100 mM Storage: Store at RT Batch Molecular Structure: 2. ANALYTICAL DATA TLC: Rf = 0.35 (Pyridine:Acetic acid:Water:Butanol [3:8:11:33]) Melting Point: Greater than 250°C 1H NMR: Consistent with structure Microanalysis: Carbon Hydrogen Nitrogen Theoretical 44.05 6.16 8.56 0 0 0 Found 43.52 6.28 8.42 0 0 0 Caution - Not Fully Tested • Research Use Only • Not For Human or Veterinary Use bio-techne.com North America China Europe Middle East Africa Rest of World [email protected] Tel: (800) 343 7475 [email protected] Tel: +44 (0)1235 529449 www.tocris.com/distributors [email protected] Tel: +86 (21) 52380373 Tel:+1 612 379 2956 Print Date: Jan 8th 2016 Product Information www.tocris.com Product Name: Guvacine hydrochloride Catalog No.: 0234 Batch No.: 13 CAS Number: 6027-91-4 IUPAC Name: 1,2,5,6-Tetrahydropyridine-3-carboxylic acid hydrochloride Description: Storage: Store at RT Specific GABA uptake inhibitor. IC values are 14, 58, 119 and 50 Solubility & Usage Info: 1870 μM for hGAT-1, rGAT-2, hGAT-3 and hBGT-1 respectively. water to 100 mM Physical and Chemical Properties: phosphate buffered saline to 100 mM Batch Molecular Formula: C H NO .HCl 69 2 Stability and Solubility Advice: Batch Molecular Weight: 163.6 Some solutions can be difficult to obtain and can be encouraged Physical Appearance: White crystalline solid by rapid stirring, sonication or gentle warming (in a 45-60°C water bath). -

Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line
Pharm Res (2018) 35: 246 https://doi.org/10.1007/s11095-018-2532-0 RESEARCH PAPER Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line Yu Takahashi1 & Tomohiro Nishimura 1 & Kei Higuchi 2 & Saki Noguchi 1 & Yuma Tega 2 & Toshiki Kurosawa2 & Yo s h i h a r u D e g u c h i 2 & Masatoshi Tomi1 Received: 14 July 2018 /Accepted: 21 October 2018 /Published online: 29 October 2018 # The Author(s) 2018 ABSTRACT Conclusions Our results indicate that LAT1, but not LAT2, Purpose The anti-epileptic drug pregabalin crosses the recognizes pregabalin as a substrate. It is suggested that LAT1 blood-brain barrier (BBB) in spite of its low lipophilicity. mediates pregabalin transport at the BBB. This study was performed to determine whether L-type amino acid transporters (LAT1/SLC7A5 and LAT2/SLC7A8) con- KEYWORDS anti-epileptic drug . blood-brain barrier . tribute to the uptake of pregabalin. L-type amino acid transporter . pregabalin Methods Pregabalin uptake by LATs-transfected HEK293 cells or hCMEC/D3 cells, an in vitro human BBB model, was measured by LC-MS/MS analysis. Expression of LAT1 mRNA in hCMEC/D3 cells was determined by quantitative ABBREVIATIONS RT-PCR analysis. BBB Blood-brain barrier Results Overexpression of LAT1, but not LAT2, in HEK293 BCH 2-Aminobicyclo-(2,2,1)-heptane-2-car- cells significantly increased the cellular uptake of pregabalin, boxylic acid GABA γ and the LAT1-mediated uptake was saturable with a Km of -Aminobutyric acid 0.288 mM. LAT1-mediated amino acid uptake was inhibited hCMEC/D3 cells Human immortalized brain endothelial specifically and almost completely in the presence of 1 mM cell line pregabalin. -

Diamandis Thesis
!"!#$ CHEMICAL GENETIC INTERROGATION OF NEURAL STEM CELLS: PHENOTYPE AND FUNCTION OF NEUROTRANSMITTER PATHWAYS IN NORMAL AND BRAIN TUMOUR INITIATING NEURAL PRECUSOR CELLS by Phedias Diamandis A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy. Department of Molecular Genetics University of Toronto © Copyright by Phedias Diamandis 2010 Phenotype and Function of Neurotransmitter Pathways in Normal and Brain Tumor Initiating Neural Precursor Cells Phedias Diamandis Doctor of Philosophy Department of Molecular Genetics University of Toronto 2010 &'(!)&*!% The identification of self-renewing and multipotent neural stem cells (NSCs) in the mammalian brain brings promise for the treatment of neurological diseases and has yielded new insight into brain cancer. The complete repertoire of signaling pathways that governs these cells however remains largely uncharacterized. This thesis describes how chemical genetic approaches can be used to probe and better define the operational circuitry of the NSC. I describe the development of a small molecule chemical genetic screen of NSCs that uncovered an unappreciated precursor role of a number of neurotransmitter pathways commonly thought to operate primarily in the mature central nervous system (CNS). Given the similarities between stem cells and cancer, I then translated this knowledge to demonstrate that these neurotransmitter regulatory effects are also conserved within cultures of cancer stem cells. I then provide experimental and epidemiologically support for this hypothesis and suggest that neurotransmitter signals may also regulate the expansion of precursor cells that drive tumor growth in the brain. Specifically, I first evaluate the effects of neurochemicals in mouse models of brain tumors. I then outline a retrospective meta-analysis of brain tumor incidence rates in psychiatric patients presumed to be chronically taking neuromodulators similar to those identified in the initial screen. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Transporters
Alexander, S. P. H., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., Armstrong, J. F., Faccenda, E., Harding, S. D., Pawson, A. J., Sharman, J. L., Southan, C., Davies, J. A., & CGTP Collaborators (2019). The Concise Guide to Pharmacology 2019/20: Transporters. British Journal of Pharmacology, 176(S1), S397-S493. https://doi.org/10.1111/bph.14753 Publisher's PDF, also known as Version of record License (if available): CC BY Link to published version (if available): 10.1111/bph.14753 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Wiley at https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14753. Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L Sharman5 , Christopher Southan5 , Jamie A Davies5 and CGTP Collaborators 1School of Life Sciences, -

O'neill2020 Redacted.Pdf (7.617Mb)
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Functional Characterisation of Spontaneously Active GABAA Receptors in Rat Dentate Gyrus Granule Cells Nathanael O’Neill B.Medsc. (Hons) Doctor of Philosophy The University of Edinburgh 2020 ii Abstract GABAA receptors (GABAARs) are the principal inhibitory neurotransmitter receptors in the adult mammalian central nervous system. GABAARs mediate two forms of inhibition: fast, phasic conductance; and slow, tonic conductance. Tonic conductance arises due to the persistent activation of GABAARs. This persistent activation can occur by GABA-dependent or GABA- independent mechanisms. Low concentrations of ambient GABA activate high affinity GABAARs located outside the synapse – at peri-/extra-synaptic sites – to generate GABA-dependent tonic conductance. In contrast, GABA-independent tonic conductance is generated by GABAARs that activate spontaneously, in the absence of GABA, due to constitutive receptor gating. -

Guvacine Hydrochloride | Medchemexpress
Inhibitors Product Data Sheet Guvacine hydrochloride • Agonists Cat. No.: HY-100809 CAS No.: 6027-91-4 Molecular Formula: C₆H₁₀ClNO₂ • Molecular Weight: 163.6 Screening Libraries Target: GABA Receptor Pathway: Membrane Transporter/Ion Channel; Neuronal Signaling Storage: Powder -20°C 3 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro H2O : 41.67 mg/mL (254.71 mM; Need ultrasonic) DMSO : 1 mg/mL (6.11 mM; ultrasonic and warming and heat to 80°C) Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 6.1125 mL 30.5623 mL 61.1247 mL Stock Solutions 5 mM 1.2225 mL 6.1125 mL 12.2249 mL 10 mM 0.6112 mL 3.0562 mL 6.1125 mL Please refer to the solubility information to select the appropriate solvent. BIOLOGICAL ACTIVITY Description Guvacine hydrochloride is an alkaloid from the nut of Areca catechu, acts as an inhibitor of GABA transporter, and dispalys modest selectivity for cloned GABA transporters with IC50s of 14 μM (human GAT-1), 39 μM (rat GAT-1), 58 μM (rat GAT-2), 119 μM (human GAT-3), 378 μM (rat GAT-3), and 1870 μM (human BGT-3). IC₅₀ & Target IC50: 14 μM (human GAT-1), 39 μM (rat GAT-1), 58 μM (rat GAT-2), 119 μM (human GAT-3), 378 μM (rat GAT-3), 1870 μM (human BGT-3)[1] In Vitro Guvacine hydrochloride is a potent inhibitor of GABA transporter, dispalys modest selectivity forcloned GABA transporters with IC50s of 14 μM (human GAT-1), 39 μM (rat GAT-1), 58 μM (rat GAT-2), 119 μM (human GAT-3), 378 μM (rat GAT-3), and [1] 1870 μM (human BGT-3). -
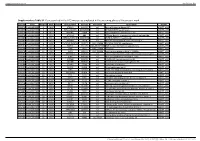
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166