Bioinformatics for Prohormone and Neuropeptide Discovery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Searching for Novel Peptide Hormones in the Human Genome Olivier Mirabeau
Searching for novel peptide hormones in the human genome Olivier Mirabeau To cite this version: Olivier Mirabeau. Searching for novel peptide hormones in the human genome. Life Sciences [q-bio]. Université Montpellier II - Sciences et Techniques du Languedoc, 2008. English. tel-00340710 HAL Id: tel-00340710 https://tel.archives-ouvertes.fr/tel-00340710 Submitted on 21 Nov 2008 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITE MONTPELLIER II SCIENCES ET TECHNIQUES DU LANGUEDOC THESE pour obtenir le grade de DOCTEUR DE L'UNIVERSITE MONTPELLIER II Discipline : Biologie Informatique Ecole Doctorale : Sciences chimiques et biologiques pour la santé Formation doctorale : Biologie-Santé Recherche de nouvelles hormones peptidiques codées par le génome humain par Olivier Mirabeau présentée et soutenue publiquement le 30 janvier 2008 JURY M. Hubert Vaudry Rapporteur M. Jean-Philippe Vert Rapporteur Mme Nadia Rosenthal Examinatrice M. Jean Martinez Président M. Olivier Gascuel Directeur M. Cornelius Gross Examinateur Résumé Résumé Cette thèse porte sur la découverte de gènes humains non caractérisés codant pour des précurseurs à hormones peptidiques. Les hormones peptidiques (PH) ont un rôle important dans la plupart des processus physiologiques du corps humain. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Regulation of Hypothalamic Signaling by Tuberoinfundibular Peptide of 39
18166 • The Journal of Neuroscience, December 7, 2011 • 31(49):18166–18179 Behavioral/Systems/Cognitive Regulation of Hypothalamic Signaling by Tuberoinfundibular Peptide of 39 Residues Is Critical for the Response to Cold: A Novel Peptidergic Mechanism of Thermoregulation Eugene L. Dimitrov, Yoon Yi Kim, and Ted B. Usdin Section On Fundamental Neuroscience, National Institute Of Mental Health, National Institutes Of Health, Bethesda, Maryland 20892 Euthermia is critical for mammalian homeostasis. Circuits within the preoptic hypothalamus regulate temperature, with fine control exerted via descending GABAergic inhibition of presympathetic motor neurons that control brown adipose tissue (BAT) thermogenesis and cutaneous vascular tone. The thermoregulatory role of hypothalamic excitatory neurons is less clear. Here we report peptidergic regulation of preoptic glutamatergic neurons that contributes to temperature regulation. Tuberoinfundibular peptide of 39 residues (TIP39) is a ligand for the parathyroid hormone 2 receptor (PTH2R). Both peptide and receptor are abundant in the preoptic hypothal- amus. Based on PTH2R and vesicular glutamate transporter 2 (VGlut2) immunolabeling in animals with retrograde tracer injection, PTH2R-containing glutamatergic fibers are presynaptic to neurons projecting from the median preoptic nucleus (MnPO) to the dorso- medial hypothalamus. Transneuronal retrograde pathway tracing with pseudorabies virus revealed connectivity between MnPO VGlut2 and PTH2R neurons and BAT. MnPO injection of TIP39 increased body temperature by 2°C for several hours. Mice lacking TIP39 signaling, either because of PTH2R-null mutation or brain delivery of a PTH2R antagonist had impaired heat production upon cold exposure, but no change in basal temperature and no impairment in response to a hot environment. Thus, TIP39 appears to act on PTH2Rs present on MnPO glutamatergic terminals to regulate their activation of projection neurons and subsequent sympathetic BAT activation. -

G Protein‐Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology (2019) 176, S21–S141 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors Stephen PH Alexander1 , Arthur Christopoulos2 , Anthony P Davenport3 , Eamonn Kelly4, Alistair Mathie5 , John A Peters6 , Emma L Veale5 ,JaneFArmstrong7 , Elena Faccenda7 ,SimonDHarding7 ,AdamJPawson7 , Joanna L Sharman7 , Christopher Southan7 , Jamie A Davies7 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University, Parkville, Victoria 3052, Australia 3Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK 4School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 5Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 6Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 7Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website. -
![SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] Product Data](https://docslib.b-cdn.net/cover/1900/six1-mouse-monoclonal-antibody-clone-id-oti1h4-product-data-1341900.webp)
SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF504057 SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] Product data: Product Type: Primary Antibodies Clone Name: OTI1H4 Applications: WB Recommended Dilution: WB 1:2000 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 1-284 of human SIX1(NP_005973) produced in E.coli. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 32 kDa Gene Name: Homo sapiens SIX homeobox 1 (SIX1), mRNA. Database Link: NP_005973 Entrez Gene 20471 MouseEntrez Gene 114634 RatEntrez Gene 6495 Human Q15475 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] – CF504057 Background: The protein encoded by this gene is a homeobox protein that is similar to the Drosophila 'sine oculis' gene product. -

The Evolution of Cell Communication: the Road Not Taken
UCLA UCLA Previously Published Works Title The Evolution of Cell Communication: The Road not Taken. Permalink https://escholarship.org/uc/item/2fx0d60p Authors Torday, JS Rehan, VK Publication Date 2009-09-01 DOI 10.4137/cci.s2776 Peer reviewed eScholarship.org Powered by the California Digital Library University of California HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Cell Commun Manuscript Author Insights. Manuscript Author manuscript; available in PMC 2015 April 15. Published in final edited form as: Cell Commun Insights. ; 2: 17–25. The Evolution of Cell Communication: The Road not Taken J.S. Torday and V.K. Rehan Department of Pediatrics, Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, Torrance, California, U.S Abstract In the post-genomic era the complex problem of evolutionary biology can be tackled from the top- down, the bottom-up, or from the middle-out. Given the emergent and contingent nature of this process, we have chosen to take the latter approach, both as a mechanistic link to developmental biology and as a rational means of identifying signaling mechanisms based on their functional genomic significance. Using this approach, we have been able to configure a working model for lung evolution by reverse-engineering lung surfactant from the mammalian lung to the swim bladder of fish. Based on this archetypal cell-molecular model, we have reduced evolutionary biology to cell communication, starting with unicellular organisms communicating with the environment, followed by cell-cell communication to generate metazoa, culminating in the communication of genetic information between generations, i.e. reproduction. This model predicts the evolution of physiologic systems-including development, homeostasis, disease, regeneration/ repair, and aging- as a logical consequence of biology reducing entropy. -

Anti-SIX1 Antibody (ARG59134)
Product datasheet [email protected] ARG59134 Package: 50 μg anti-SIX1 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes SIX1 Tested Reactivity Hu Tested Application WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name SIX1 Antigen Species Human Immunogen Synthetic peptide corresponding to aa. 245-282 of Human SIX1. (NYSLPGLTASQPSHGLQTHQHQLQDSLLGPLTSSLVDL) Conjugation Un-conjugated Alternate Names Sine oculis homeobox homolog 1; Homeobox protein SIX1; DFNA23; TIP39; BOS3 Application Instructions Application table Application Dilution WB 0.1 - 0.5 µg/ml Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 32 kDa Properties Form Liquid Purification Affinity purification with immunogen. Buffer 0.9% NaCl, 0.2% Na2HPO4, 0.05% Sodium azide and 5% BSA. Preservative 0.05% Sodium azide Stabilizer 5% BSA Concentration 0.5 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C or below. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol SIX1 Gene Full Name SIX homeobox 1 Background The protein encoded by this gene is a homeobox protein that is similar to the Drosophila 'sine oculis' gene product. This gene is found in a cluster of related genes on chromosome 14 and is thought to be involved in limb development. -

Characterization of the Prohormone Complement in Cattle Using Genomic
BMC Genomics BioMed Central Research article Open Access Characterization of the prohormone complement in cattle using genomic libraries and cleavage prediction approaches Bruce R Southey*1,2, Sandra L Rodriguez-Zas2 and Jonathan V Sweedler1 Address: 1Department of Chemistry, University of Illinois, Urbana, IL, USA and 2Department of Animal Sciences, University of Illinois, Urbana IL, USA Email: Bruce R Southey* - [email protected]; Sandra L Rodriguez-Zas - [email protected]; Jonathan V Sweedler - [email protected] * Corresponding author Published: 16 May 2009 Received: 10 December 2008 Accepted: 16 May 2009 BMC Genomics 2009, 10:228 doi:10.1186/1471-2164-10-228 This article is available from: http://www.biomedcentral.com/1471-2164/10/228 © 2009 Southey et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Neuropeptides are cell to cell signalling molecules that regulate many critical biological processes including development, growth and reproduction. These peptides result from the complex processing of prohormone proteins, making their characterization both challenging and resource demanding. In fact, only 42 neuropeptide genes have been empirically confirmed in cattle. Neuropeptide research using high-throughput technologies such as microarray and mass spectrometry require accurate annotation of prohormone genes and products. However, the annotation and associated prediction efforts, when based solely on sequence homology to species with known neuropeptides, can be problematic. Results: Complementary bioinformatic resources were integrated in the first survey of the cattle neuropeptide complement. -

Strong Linkage on 2Q33.3 to Familial Early-Onset Generalized Osteoarthritis and a Consideration of Two Positional Candidate Genes
European Journal of Human Genetics (2006) 14, 1280–1287 & 2006 Nature Publishing Group All rights reserved 1018-4813/06 $30.00 www.nature.com/ejhg ARTICLE Strong linkage on 2q33.3 to familial early-onset generalized osteoarthritis and a consideration of two positional candidate genes Ingrid Meulenbelt*,1, Josine L Min1, Cornelia M van Duijn4, Margreet Kloppenburg2,3, Ferdinand C Breedveld2 and P Eline Slagboom1 1Molecular Epidemiology, Leiden University Medical Center, Leiden, The Netherlands; 2Department of Rheumatology, Leiden University Medical Center, Leiden, The Netherlands; 3Departmentof Clinical Epidemiology, Leiden University Medical Center, Leiden, The Netherlands; 4Department of Epidemiology and Biostatistics Erasmus University Medical School, Rotterdam, The Netherlands A genomewide screen was performed in four extended families with early-onset generalized osteoarthritis (FOA) without dysplasia. The FOA phenotype within these families shows a dominant Mendelian inheritance pattern and may represent common osteoarthritis (OA) at later ages. An initial locus was confirmed by three additional families and refined by 14 markers to a two-point logarithm of odds score of 6.05 (theta ¼ 0.00) for marker D2S155 at chromosome 2q33.3. This locus coincided with the highest multipoint nonparametric linkage score of 4.70 (P-value ¼ 0.0013) at marker D2S2358. Haplotype analysis of family members delineated a narrow region with a number of possible positional candidates, of which we investigate here the two most likely ones: PTHR2, encoding parathyroid hormone receptor 2, and FZD5, encoding frizzled receptor 5. For FZD5, we did not observe a segregating variant, however, for PTHR2,a missense variant (A225S) cosegregated with FOA in one family. The frequency of the PTHR2 variant was rare in a population-based sample, aged 55–70 years (N ¼ 1228, 0.4%). -
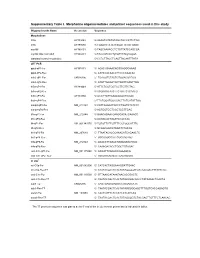
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Functional Characterization and Evolution of PTH/Pthrp Receptors
Pinheiro et al. BMC Evolutionary Biology 2012, 12:110 http://www.biomedcentral.com/1471-2148/12/110 RESEARCH ARTICLE Open Access Functional characterization and evolution of PTH/PTHrP receptors: insights from the chicken Pedro LC Pinheiro†, João CR Cardoso*†, Deborah M Power and Adelino V M Canário Abstract Background: The parathyroid hormone (PTH)-family consists of a group of structurally related factors that regulate calcium and bone homeostasis and are also involved in development of organs such as the heart, mammary gland and immune system. They interact with specific members of family 2 B1 G-protein coupled receptors (GPCRs), which have been characterised in teleosts and mammals. Two PTH/PTHrP receptors, PTH1R and PTH2R exist in mammals and in teleost fish a further receptor PTH3R has also been identified. Recently in chicken, PTH-family members involved in calcium transport were characterized and specific PTHRs are suggested to exist although they have not yet been isolated or functionally characterized. The aim of this study is to further explore the evolution and function of the vertebrate PTH/PTHrP system through the isolation, phylogenetic analysis and functional characterization of the chicken receptors. Results: Two PTHRs were isolated in chicken and sequence comparison and phylogenetic analysis indicate that the chicken receptors correspond to PTH1R and PTH3R, which emerged prior to the teleost/tetrapod divergence since they are present in cartilaginous fish. The vertebrate PTH2R receptor and its ligand TIP39 have been lost from bird genomes. Chicken PTH1R and PTH3R have a divergent and widespread tissue expression and are also evident in very early embryonic stages of development. -

TA504057 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA504057 SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] Product data: Product Type: Primary Antibodies Clone Name: OTI1H4 Applications: WB Recommend Dilution: WB 1:2000 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 1-284 of human SIX1(NP_005973) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 0.77 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Predicted Protein Size: 32 kDa Gene Name: SIX homeobox 1 Database Link: NP_005973 Entrez Gene 6495 Human Background: The protein encoded by this gene is a homeobox protein that is similar to the Drosophila 'sine oculis' gene product. This gene is found in a cluster of related genes on chromosome 14 and is thought to be involved in limb development. Defects in this gene are a cause of autosomal dominant deafness type 23 (DFNA23) and branchiootic syndrome type 3 (BOS3). [provided by RefSeq] Synonyms: BOS3; DFNA23; TIP39 Protein Families: Transcription Factors This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2020 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 SIX1 Mouse Monoclonal Antibody [Clone ID: OTI1H4] – TA504057 Product images: HEK293T cells were transfected with the pCMV6- ENTRY control (Left lane) or pCMV6-ENTRY SIX1 ([RC203465], Right lane) cDNA for 48 hrs and lysed.