A Comprehensive Kinetic Model of Ethylene and Acetylene Oxidation at High Temperatures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Use of Solvents for Pahs Extraction and Enhancement of the Pahs Bioremediation in Coal- Tar-Contaminated Soils Pak-Hing Lee Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2000 Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal- tar-contaminated soils Pak-Hing Lee Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Environmental Engineering Commons Recommended Citation Lee, Pak-Hing, "Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal-tar-contaminated soils " (2000). Retrospective Theses and Dissertations. 13912. https://lib.dr.iastate.edu/rtd/13912 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter fece, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quaiity of the copy submitted. Broken or indistinct print colored or poor quality illustrations and photographs, print bleedthrough, substeindard margins, and improper alignment can adversely affect reproduction. In the unlilcely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -

Ethylene Dichloride (Edc) Handbook
ETHYLENE DICHLORIDE (EDC) HANDBOOK OXYCHEM TECHNICAL INFORMATION 11/2014 Dallas-based Occidental Chemical Corporation is a leading North American manufacturer of basic chemicals, vinyls and performance chemicals directly and through various affiliates (collectively, OxyChem). OxyChem is also North America's largest producer of sodium chlorite. As a Responsible Care® company, OxyChem's global commitment to safety and the environment goes well beyond compliance. OxyChem's Health, Environment and Safety philosophy is a positive motivational force for our employees, and helps create a strong culture for protecting human health and the environment. Our risk management programs and methods have been, and continue to be, recognized as some of the industry's best. OxyChem offers an effective combination of industry expertise, experience, on line business tools, quality products and exceptional customer service. As a member of the Occidental Petroleum Corporation family, OxyChem represents a rich history of experience, top-notch business acumen, and sound, ethical business practices. 1 Table of Contents Page Introduction to Ethylene Dichloride ............................................................................................................ 3 Manufacturing .................................................................................................................................................. 3 Ethylene Dichloride (EDC) — Uses ................................................................................................................ -
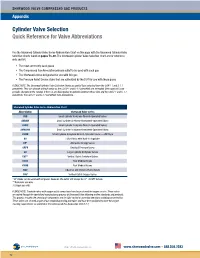
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Toxicological Profile for Ethylbenzene
ETHYLBENZENE 151 5. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL 5.1 PRODUCTION Ethylbenzene is primarily produced by the alkylation of benzene with ethylene in liquid-phase slurry reactors promoted with aluminum chloride catalysts or by vapor-phase reaction of benzene with dilute ethylene-containing feedstock with a boron trifluoride catalyst supported on alumina (Cannella 2007; Clayton and Clayton 1981; HSDB 2009; Welch et al. 2005; Ransley 1984). Newer versions of the method employ synthetic zeolites in fixed-bed reactors as catalysts for alkylation in the liquid phase or narrow pore synthetic zeolites in fixed-bed reactors in the vapor phase (Welch et al. 2005). Other methods of manufacturing ethylbenzene include preparation from acetophenone, dehydrogenation of naphthenes, catalytic cyclization and aromatization, separation from mixed xylenes via fractionation, reaction of ethylmagnesium bromide and chlorobenzene, extraction from coal oil, and recovery from benzene-toluene-xylene (BTX) processing(Clayton and Clayton 1981; HSDB 2009; Ransley 1984; Welch et al. 2005). Commercial grades of ethylbenzene may contain small amounts of m-xylene, p-xylene, cumene, and toluene (HSDB 2009). Ethylbenzene is traditionally ranked as one of the top 50 chemicals produced in the United States. Table 5-1 shows the historical production volumes of ethylbenzene from 1983 to 2005 (C&EN 1994a, 1994b, 1995, 2006; Kirschner 1995). Table 5-2 lists the facilities in each state that manufacture or process ethylbenzene, the intended use, and the range of maximum amounts of ethylbenzene that are stored on site. There are currently 3,755 facilities that produce, process, or use ethylbenzene in the United States. The data listed in Table 5-2 are derived from the Toxics Release Inventory (TRI06 2008). -

ETHYLENE from METHANE (January 1994)
Abstract Process Economics Program Report No. 208 ETHYLENE FROM METHANE (January 1994) This report evaluates two routes for the production of ethylene from methane: the direct synthesis based on the oxidative coupling of methane, and the less direct chemistry of converting methanol (which is derived from methane via synthesis gas) in the presence of an aluminophosphate molecular sieve catalyst. Our evaluations indicate that at the present state of development, the economics of both routes are unattractive when compared with the steam pyrolysis of hydrocarbons. We analyze the results of our evaluations to define the technical targets that must be attained for success. We also present a comprehensive technical review that examines not only the two routes evaluated, but also some of the more promising alternative approaches, such as synthesis gas conversion via a modified Fischer-Tropsch process, ethanol synthesis by the homologation of methanol, and ethylene production via methyl chloride. This report will be of interest to petrochemical companies that produce or consume ethylene and to energy-based companies (or equivalent government organizations in various countries) that have access to or control large resources of methane-rich natural gas. PEP’91 SCN CONTENTS 1 INTRODUCTION 1-1 2 SUMMARY 2-1 TECHNICAL REVIEW 2-1 Oxidative Coupling 2-1 Methanol Conversion to Ethylene 2-3 Modified Fischer-Tropsch (FT) Process 2-3 Methanol Homologation 2-3 Conversion via Methyl Chloride 2-4 SRI’S PROCESS CONCEPTS 2-4 Ethylene from Methane by Oxidative -

Hydrogenation of Ethylene on Metallic Catalysts
S ro Hating Bure* M “"“^ piu&« Ubwu, Ml ®min’ JUN 2 1 1S68 A 1 1 1 2 mbESD NATX INST OF STANDARDS & TECH R.I.C. NSRDS-NBS 13 NSRDS 11 021 46250 ™SRDS.NB^ QC100 -U573 V13;1968 C.1 sH *- NBS-PUB-C 1964 ^f#Cf DftU NBS 'USUCATfONS Hydrogenation of Ethylene Metallic Catalysts U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS National Standard Reference Data Series National Bureau of Standards National Standard Reference Data System, Plan of Operation, NSRDS-NBS 1 — 15 cents* Thermal Properties of Aqueous Uni-univalent Electrolytes NSRDS-NBS 2 — 45 cents* Selected Tables of Atomic Spectra, Atomic Energy Levels and Multiplet Tables — Si II, Si ill, Si iv, NSRDS-NBS 3, Section 1—35 cents* Selected Tables of Atomic Spectra, Atomic Energy Levels and Multiplet Tables — Si I NSRDS — NBS 3, Section 2 — 20 cents* Atomic Transition Probabilities, Volume I, Hydrogen Through Neon, NSRDS-NBS 4 — $2.50* The Band Spectrum of Carbon Monoxide, NSRDS-NBS 5 — 70 cents* Tables of Molecular Vibrational Frequencies. Part 1, NSRDS-NBS 6 — 40 cents* High Temperature Properties and Decomposition of Inorganic Salts. Part 1. Sulfates, NSRDS-NBS 7-35 cents* Thermal Conductivity of Selected Materials, NSRDS-NBS 8 — $1.00* Tables of Biomolecular Gas Reactions, NSRDS-NBS 9 — $2.00* Selected Values of Electric Dipole Moments for Molecules in the Gas Phase, NSRDS- NBS 10 — 40 cents* Tables of Molecular Vibrational Frequencies, Part 2, NSRDS-NBS 11 — 30 cents* Tables for the Rigid Asymmetric Rotor: Transformation Coefficient from Symmetric to Asymmetric Bases Expectation Values of P\ and 4 NSRDS-NBS 12 — in press. -

Acetylene and Ethylene Hydrogenation on Alumina Supported Pd-Ag Model Catalysts
Catalysis Letters Vol. 108, Nos. 3–4, May 2006 (Ó 2006) 159 DOI: 10.1007/s10562-006-0041-y Acetylene and ethylene hydrogenation on alumina supported Pd-Ag model catalysts N.A. Khan,* S. Shaikhutdinov, and H.-J. Freund Department of Chemical Physics, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, Berlin 14195, Germany Received 20 December 2005; accepted 20 January 2006 Adsorption and co-adsorption of ethylene, acetylene and hydrogen on Pd-Ag particles, supported on thin alumina films, have been studied by temperature programmed desorption (TPD). The TPD results show that adding of Ag to Pd suppresses overall hydrogenation activity but increases selectivity towards ethylene, i.e. similar to that observed on real catalysts. The results are rationalized on the basis of a complex interplay between surface and subsurface hydrogen species available in the system, whereby the latter species are the most critical for total hydrogenation of acetylene to ethane. KEY WORDS: hydrogenation; bimetallic catalysts; acetylene; palladium; silver. The selective hydrogenation of acetylene is an alumina film both at low pressures (with TPD) and high industrially important catalytic process in the large-scale pressures (up to 1 bar, using gas chromatography [16– production of polyethylene, where a small quantity of 18]. Under both conditions, the ethylene hydrogenation acetylene (<3%) is present in ethylene feedstock. reaction was found to be structure insensitive. In Commercially, it is preferred to reduce the acetylene contrast, hydrogenation of 2-pentenes exhibited a sig- content to less than 10 ppm, which needs 99% acet- nificant particle size effect [19]. These studies have ylene conversion in the excess of ethylene [1]. -

Guidelines for Drinking-Water Quality, Fourth Edition
12 Chemical fact sheets A conceptual framework for Introduction implementing the Guidelines (Chapter 1) (Chapter 2) he background docu- FRAMEWORK FOR SAFE DRINKING-WATER SUPPORTING Tments referred to in INFORMATION this chapter (as the princi- Health-based targets Public health context Microbial aspects pal reference for each fact (Chapter 3) and health outcome (Chapters 7 and 11) sheet) may be found on Water safety plans Chemical aspects (Chapter 4) (Chapters 8 and 12) the Water, Sanitation, Hy- System Management and Radiological Monitoring giene and Health web site assessment communication aspects at http://www.who.int/ (Chapter 9) Acceptability Surveillance water_sanitation_health/ aspects (Chapter 5) dwq/chemicals/en/index. (Chapter 10) html. A complete list of r eferences cited in this Application of the Guidelines in specic circumstances chapter, including the (Chapter 6) background documents Climate change, Emergencies, Rainwater harvesting, Desalination for each c hemical, is pro- systems, Travellers, Planes and vided in Annex 2. ships, etc. 12.1 Chemical contaminants in drinking-water Acrylamide Residual acrylamide monomer occurs in polyacrylamide coagulants used in the treat- ment of drinking-water. In general, the maximum authorized dose of polymer is 1 mg/l. At a monomer content of 0.05%, this corresponds to a maximum theoretical concen- tration of 0.5 µg/l of the monomer in water. Practical concentrations may be lower by a factor of 2–3. This applies to the anionic and non-ionic polyacrylamides, but residual levels from cationic polyacrylamides may be higher. Polyacrylamides are also used as grouting agents in the construction of drinking-water reservoirs and wells. -

Kinetic Study of the Selective Hydrogenation of Acetylene Over Supported Palladium Under Tail-End Conditions
catalysts Article Kinetic Study of the Selective Hydrogenation of Acetylene over Supported Palladium under Tail-End Conditions Caroline Urmès 1,2, Jean-Marc Schweitzer 2, Amandine Cabiac 2 and Yves Schuurman 1,* 1 IRCELYON CNRS, UMR 5256, Univ Lyon, Université Claude Bernard Lyon 1, 2 avenue Albert Einstein, 69626 Villeurbanne Cedex, France; [email protected] 2 IFP Energies nouvelles, Etablissement de Lyon, Rond-point de l’échangeur de Solaize, BP3, 69360 Solaize, France; [email protected] (J.-M.S.); [email protected] (A.C.) * Correspondence: [email protected]; Tel.: +33-472445482 Received: 9 January 2019; Accepted: 31 January 2019; Published: 14 February 2019 Abstract: The kinetics of the selective hydrogenation of acetylene in the presence of an excess of ethylene has been studied over a 0.05 wt. % Pd/α-Al2O3 catalyst. The experimental reaction conditions were chosen to operate under intrinsic kinetic conditions, free from heat and mass transfer limitations. The data could be described adequately by a Langmuir–Hinshelwood rate-equation based on a series of sequential hydrogen additions according to the Horiuti–Polanyi mechanism. The mechanism involves a single active site on which both the conversion of acetylene and ethylene take place. Keywords: power-law; Langmuir–Hinshelwood; kinetic modeling; Pd/α-Al2O3 1. Introduction Ethylene is the largest of the basic chemical building blocks with a global market estimated at more than 140 million tons per year with an increasing growth rate. It is used mainly as precursor for polymers production, for instance polyethylene, vinyl chloride, ethylbenzene, or even ethylene oxide synthesis. -

A DFT Study of Synthesis of Acetic Acid from Methane and Carbon Dioxide
_______________________________________________________________________________www.paper.edu.cn Chemical Physics Letters 368 (2003) 313–318 www.elsevier.com/locate/cplett A DFT study of synthesis of acetic acid from methane and carbon dioxide Jian-guo Wang a, Chang-jun Liu a,*, Yue-ping Zhang b, Baldur Eliasson c a State Key Laboratory of C1 Chemistry and Technology, ABB Plasma Greenhouse Gas Chemistry Laboratory and School of Chemical Engineering, Tianjin University, Tianjin 300072, PR China b Department of Chemistry, Tianjin University, Tianjin 300072, PR China c ABB Switzerland Ltd., CH5405, Baden, Switzerland Received 2 October 2002; in final form 20 November 2002 Abstract We have previously reported an experimental investigation on synthesis of acetic acid directly from CH4 and CO2 via dielectric-barrier discharge. In this work, a DFT study was conducted using three hybrid DFT methods in order to À understand the mechanism of such direct synthesis. It suggests that the synthesis is via two pathways with CO2 and CO À as key intermediates. The energy requirement with CO2 pathway is much less than that with CO. The methyl radical formation and the dissociation of CO2 are two limiting steps for the synthesis of acetic acid directly from CH4 and CO2. Ó 2002 Elsevier Science B.V. All rights reserved. 1. Introduction [1,2]. But methane utilization still remains as a big challenge to chemists all over the world. Methane is the principal component of natural One of the important target molecules of direct gas, coalbed methane, associated gas of oil fields methane conversion is acetic acid [5–9] and some by-product gases of chemical plants. -

Ijmp.Jor.Br V
INDEPENDENT JOURNAL OF MANAGEMENT & PRODUCTION (IJM&P) http://www.ijmp.jor.br v. 10, n. 8, Special Edition Seng 2019 ISSN: 2236-269X DOI: 10.14807/ijmp.v10i8.1046 A NEW HYPOTHESIS ABOUT THE NUCLEAR HYDROGEN STRUCTURE Relly Victoria Virgil Petrescu IFToMM, Romania E-mail: [email protected] Raffaella Aversa University of Naples, Italy E-mail: [email protected] Antonio Apicella University of Naples, Italy E-mail: [email protected] Taher M. Abu-Lebdeh North Carolina A and T State Univesity, United States E-mail: [email protected] Florian Ion Tiberiu Petrescu IFToMM, Romania E-mail: [email protected] Submission: 5/3/2019 Accept: 5/20/2019 ABSTRACT In other papers already presented on the structure and dimensions of elemental hydrogen, the elementary particle dynamics was taken into account in order to be able to determine the size of the hydrogen. This new work, one comes back with a new dynamic hypothesis designed to fundamentally change again the dynamic particle size due to the impulse influence of the particle. Until now it has been assumed that the impulse of an elementary particle is equal to the mass of the particle multiplied by its velocity, but in reality, the impulse definition is different, which is derived from the translational kinetic energy in a rapport of its velocity. This produces an additional condensation of matter in its elemental form. Keywords: Particle structure; Impulse; Condensed matter. [http://creativecommons.org/licenses/by/3.0/us/] Licensed under a Creative Commons Attribution 3.0 United States License 1749 INDEPENDENT JOURNAL OF MANAGEMENT & PRODUCTION (IJM&P) http://www.ijmp.jor.br v.