Kinetic Study of the Selective Hydrogenation of Acetylene Over Supported Palladium Under Tail-End Conditions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Production of Cyclohexane Through Catalytic Hydrogenation of Benzene
Production of Cyclohexane through Catalytic Hydrogenation of Benzene Background Cyclohexane is industrially produced from Benzene as it is not a naturally available resource. Cyclohexane undergoes oxidation reactions yielding Cyclohexanone and Cyclohexanol which are precursors for the production of Adipic acid and Caprolactum. Caprolactum is the raw material used for producing polymer Nylon-6. Benzene reacts with a mixture of hydrogen and methane in contact with a Nickel based catalyst producing Cyclohexane. The conversion of this vapour phase reaction is almost 99%. Reaction involved: Benzene + Hydrogen Cyclohexane (Vapour Phase) Reactor Used: Catalytic Packed Bed Conversion Reactor Reactor conditions: Outlet Temperature = 497 K, Pressure Drop = 1.02 atm Catalyst Used: Nickel Based Process Description Fresh benzene (370 kmol/h) and excess hydrogen (1470 kmol/h) is preheated to a temperature of 422 K and sent to a packed bed reactor. A vapour phase reaction occurs in the reactor at 497 K which converts benzene to cyclohexane through catalytic hydrogenation of benzene. The conversion of this reaction is about 99%. The reactor products are cooled to 370 K and sent through a pressure reduction valve which reduces the pressure of the stream from 30 atm to 24 atm. A two stage separator separates the product cyclohexane from unreacted hydrogen and methane- first at a high pressure (24 atm) and then at a lower pressure (3 atm). The unreacted hydrogen-methane mixture is recovered from the top of the flash column and is sent to a splitter having a splittling ratio of 9:1. The smaller stream is sent as a recycle stream and mixes with fresh hydrogen, while the rest is drawn out as fuel gas for incinerators. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -

Appendix F. Glossary
Appendix F. Glossary 2DEG 2-dimensional electron gas A/D Analog to digital AAAR American Association for Aerosol Research ADC Analog-digital converter AEM Analytical electron microscopy AFM Atomic force microscope/microscopy AFOSR Air Force Office of Scientific Research AIST (Japan) Agency of Industrial Science and Technology AIST (Japan, MITI) Agency of Industrial Science and Technology AMLCD Active matrix liquid crystal display AMM Amorphous microporous mixed (oxides) AMO Atomic, molecular, and optical AMR Anisotropic magnetoresistance ARO (U.S.) Army Research Office ARPES Angle-resolved photoelectron spectroscopy ASET (Japan) Association of Super-Advanced Electronics Technologies ASTC Australia Science and Technology Council ATP (Japan) Angstrom Technology Partnership ATP Adenosine triphosphate B Magnetic flux density B/H loop Closed figure showing B (magnetic flux density) compared to H (magnetic field strength) in a magnetizable material—also called hysteresis loop bcc Body-centered cubic BMBF (Germany) Ministry of Education, Science, Research, and Technology (formerly called BMFT) BOD-FF Bond-order-dependent force field BRITE/EURAM Basic Research of Industrial Technologies for Europe, European Research on Advanced Materials program CAD Computer-assisted design CAIBE Chemically assisted ion beam etching CBE Chemical beam epitaxy 327 328 Appendix F. Glossary CBED Convergent beam electron diffraction cermet Ceramic/metal composite CIP Cold isostatic press CMOS Complementary metal-oxide semiconductor CMP Chemical mechanical polishing -

Ethylene Dichloride (Edc) Handbook
ETHYLENE DICHLORIDE (EDC) HANDBOOK OXYCHEM TECHNICAL INFORMATION 11/2014 Dallas-based Occidental Chemical Corporation is a leading North American manufacturer of basic chemicals, vinyls and performance chemicals directly and through various affiliates (collectively, OxyChem). OxyChem is also North America's largest producer of sodium chlorite. As a Responsible Care® company, OxyChem's global commitment to safety and the environment goes well beyond compliance. OxyChem's Health, Environment and Safety philosophy is a positive motivational force for our employees, and helps create a strong culture for protecting human health and the environment. Our risk management programs and methods have been, and continue to be, recognized as some of the industry's best. OxyChem offers an effective combination of industry expertise, experience, on line business tools, quality products and exceptional customer service. As a member of the Occidental Petroleum Corporation family, OxyChem represents a rich history of experience, top-notch business acumen, and sound, ethical business practices. 1 Table of Contents Page Introduction to Ethylene Dichloride ............................................................................................................ 3 Manufacturing .................................................................................................................................................. 3 Ethylene Dichloride (EDC) — Uses ................................................................................................................ -
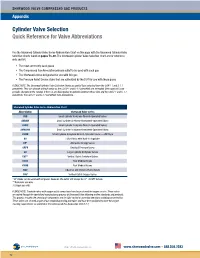
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Chapter 3 CHEMICAL MODIFICATION of OILS and FATS
Chapter 3 CHEMICAL MODIFICATION OF OILS AND FATS From the fats and oils obtained from natural resources, the majority of them are used directly or just after refinement. While the others are used after modification by chemical process. This chapter lists some typical modifications of fats and oils by chemical means. 3-1 Alkaline Hydrolysis Figure 3-1-1: Alkaline hydrolysis (saponification) of oil to make soaps. There are many methods for hydrolysis of triacylglycerol molecule. The most common method is alkaline hydrolysis. Heating (around 100˚C)triacylglycerols with aqueous solu- tion of sodium hydroxide results in glycerol and alkaline salt of fatty acid (i.e. soap) (Figure 3-1-1). This is called saponification, and used for production of soap. 3-2 Hydrogenation Number of double bonds in oils and fats affects physical property such as melting point, crystallinity. Generally, double bonds reduce the oil’s melting point. Therefore, oils rich in unsaturated fatty acids are liquid, while ones with small amount of unsaturated fatty acids are solid or semi-solid. Hydrogenation is a process to add hydrogen atoms into double bonds of unsaturated fatty acids (Figure 3-2-1). As the result of hydrogenation, liquid oil becomes solid or semi-solid. A typical example of hydrogenation is in the process of margarine and shortening production. Vegetable oil is hydrogenated with gaseous H2 in the presence of a metal catalyst (usually nickel catalyst). If the hydrogenation is completely performed, all the double bounds are 19 Figure 3-2-1: Hydrogenation. converted to the saturated ones with the same carbon number. -

Hydrogenation Reaction
SOP: How to Run an Atmospheric-Pressure Hydrogenation Reaction Hazards: Hydrogenation reactions pose a significant fire hazard due to the use of flammable reagents and solvents. Such reagents include palladium on carbon (Pd/C), which is highly flammable and can ignite solvents and hydrogen. It is especially dangerous after having been used for the hydrogenation. The presence of hydrogen gas increases the risk of explosion. Special Precautions: Remove any excess clutter and any flammable solvents that are not needed from your fume hood for the reaction. Be prepared for the possibility of a small fire. Do not panic if this occurs, but simply cover the flask or funnel in which there is a fire with a watch glass and it will go out. Have a suitable sized watch glass on hand. Recommended Apparatus: A three-necked flask equipped with a magnetic stirring bar, a nitrogen inlet adapter connected to a nitrogen/vacuum manifold, a glass stopper or rubber septum, and a gas inlet adapter with a stopcock and a balloon filled with hydrogen. Procedure: 1. Put a weighed quantity of the catalyst in the flask. 2. Evacuate and back-fill the flask with nitrogen 3 times. 3. Add your solvent under a countercurrent of nitrogen. CAUTION: Do not pour your solvent from a 4-liter bottle or a 1-liter bottle. Use a small Erlenmeyer flask (for example 125 mL) containing only the needed amount of solvent. 4. Add your substrate to be hydrogenated to the flask. 5. Evacuate and back-fill the flask with hydrogen. 6. If needed, you may replace the balloon with a full one as needed during the reaction. -

Toxicological Profile for Ethylbenzene
ETHYLBENZENE 151 5. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL 5.1 PRODUCTION Ethylbenzene is primarily produced by the alkylation of benzene with ethylene in liquid-phase slurry reactors promoted with aluminum chloride catalysts or by vapor-phase reaction of benzene with dilute ethylene-containing feedstock with a boron trifluoride catalyst supported on alumina (Cannella 2007; Clayton and Clayton 1981; HSDB 2009; Welch et al. 2005; Ransley 1984). Newer versions of the method employ synthetic zeolites in fixed-bed reactors as catalysts for alkylation in the liquid phase or narrow pore synthetic zeolites in fixed-bed reactors in the vapor phase (Welch et al. 2005). Other methods of manufacturing ethylbenzene include preparation from acetophenone, dehydrogenation of naphthenes, catalytic cyclization and aromatization, separation from mixed xylenes via fractionation, reaction of ethylmagnesium bromide and chlorobenzene, extraction from coal oil, and recovery from benzene-toluene-xylene (BTX) processing(Clayton and Clayton 1981; HSDB 2009; Ransley 1984; Welch et al. 2005). Commercial grades of ethylbenzene may contain small amounts of m-xylene, p-xylene, cumene, and toluene (HSDB 2009). Ethylbenzene is traditionally ranked as one of the top 50 chemicals produced in the United States. Table 5-1 shows the historical production volumes of ethylbenzene from 1983 to 2005 (C&EN 1994a, 1994b, 1995, 2006; Kirschner 1995). Table 5-2 lists the facilities in each state that manufacture or process ethylbenzene, the intended use, and the range of maximum amounts of ethylbenzene that are stored on site. There are currently 3,755 facilities that produce, process, or use ethylbenzene in the United States. The data listed in Table 5-2 are derived from the Toxics Release Inventory (TRI06 2008). -

Catalytic Hydrogenation and Dehydrogenation Reactions of N-Alkyl-Bis(Carbazole)-Based Hydrogen Storage Materials
catalysts Article Catalytic Hydrogenation and Dehydrogenation Reactions of N-alkyl-bis(carbazole)-Based Hydrogen Storage Materials Joori Jung 1,2,†, Byeong Soo Shin 3,4,†, Jeong Won Kang 4,5,* and Won-Sik Han 1,* 1 Department of Chemistry, Seoul Women’s University, Seoul 01797, Korea; [email protected] 2 REYON Pharmaceutical, Co., Ltd., Anyang 01459, Korea 3 Hyundai Motor Company, Strategy & Technology Division, Ulwang 16082, Korea; [email protected] 4 Department of Chemical and Biological Engineering, Korea University, Seoul 02841, Korea 5 Graduate School of Energy and Environment, Korea University, Seoul 02841, Korea * Correspondence: [email protected] (J.W.K.); [email protected] (W.-S.H.) † These authors contributed equally. Abstract: Recently, there have been numerous efforts to develop hydrogen-rich organic materials because hydrogen energy is emerging as a renewable energy source. In this regard, we designed and prepared four new materials based on N-alkyl-bis(carbazole), 9,90-(2-methylpropane-1,3-diyl)bis(9H- carbazole) (MBC), 9,90-(2-ethylpropane-1,3-diyl)bis(9H-carbazole) (EBC), 9,90-(2-propylpropane-1,3- diyl)bis(9H-carbazole) (PBC), and 9,90-(2-butylpropane-1,3-diyl)bis(9H-carbazole) (BBC), to investi- gate their hydrogen adsorption/hydrogen desorption reactivity depending on the length of the alkyl chain. The gravimetric densities of MBC, EBC, PBC, and BBC were 5.86, 5.76, 5.49, and 5.31 H2 wt %, respectively, again depending on the alkyl chain length. All materials showed complete hydro- genation reactions under ruthenium on an alumina catalyst at 190 ◦C, and complete reverse reactions ◦ and dehydrogenation reactions were observed under palladium on an alumina catalyst at <280 C. -

ETHYLENE from METHANE (January 1994)
Abstract Process Economics Program Report No. 208 ETHYLENE FROM METHANE (January 1994) This report evaluates two routes for the production of ethylene from methane: the direct synthesis based on the oxidative coupling of methane, and the less direct chemistry of converting methanol (which is derived from methane via synthesis gas) in the presence of an aluminophosphate molecular sieve catalyst. Our evaluations indicate that at the present state of development, the economics of both routes are unattractive when compared with the steam pyrolysis of hydrocarbons. We analyze the results of our evaluations to define the technical targets that must be attained for success. We also present a comprehensive technical review that examines not only the two routes evaluated, but also some of the more promising alternative approaches, such as synthesis gas conversion via a modified Fischer-Tropsch process, ethanol synthesis by the homologation of methanol, and ethylene production via methyl chloride. This report will be of interest to petrochemical companies that produce or consume ethylene and to energy-based companies (or equivalent government organizations in various countries) that have access to or control large resources of methane-rich natural gas. PEP’91 SCN CONTENTS 1 INTRODUCTION 1-1 2 SUMMARY 2-1 TECHNICAL REVIEW 2-1 Oxidative Coupling 2-1 Methanol Conversion to Ethylene 2-3 Modified Fischer-Tropsch (FT) Process 2-3 Methanol Homologation 2-3 Conversion via Methyl Chloride 2-4 SRI’S PROCESS CONCEPTS 2-4 Ethylene from Methane by Oxidative -

Catalytic Hydrogenation Syringe Pump Application Note Using Teledyne Isco Syringe Pumps AN19
Catalytic Hydrogenation Syringe Pump Application Note Using Teledyne Isco Syringe Pumps AN19 Overview Methodology Hydrogenation is a chemical reaction of great impor- Molecular hydrogen does not readily react with tance to the petrochemical and fine chemical industries. organic molecules; a catalyst is always required. A cata- In its most elementary sense, the term hydrogenation lyst is a substance that controls a chemical reaction, but refers to the addition reaction of molecular hydrogen is not consumed or part of the final product. Catalysts with an unsaturated carbon-carbon double bond as work by lowering the activation energy needed for col- illustrated: liding molecules to reach the transition state. Therefore, catalysts can allow reactions to take place that would otherwise not be possible or allow them to happen at a HH HH much faster rate. A comparison of the effect catalysts can have in the + energy required can be seen in Figure 2: CC H2 CC HH energy Figure 1: Typical hydrogen reaction The first compound, called an alkene, is converted into the corresponding alkane. activation In addition to the example above, there are other energy ways in which molecular hydrogen can be reacted with activation other types of molecules. These include the incorpora- energy tion of hydrogen accompanied by cleavage of the starting molecule as in hydrodesulfurization and reac- tions in which the starting molecule undergoes rearrangement such as isomerization. uncatalysed reaction Hydrogenation in Practical Use catalysed reaction time For the petrochemical industry, many of the com- pounds found in crude oil are of little use since they Figure 2: Boltzman Energy Diagram for reaction contain multiple double bonds; they must be first con- pathway verted to saturated compounds before use as commodities such as gasoline. -

Hydrogenation of Ethylene on Metallic Catalysts
S ro Hating Bure* M “"“^ piu&« Ubwu, Ml ®min’ JUN 2 1 1S68 A 1 1 1 2 mbESD NATX INST OF STANDARDS & TECH R.I.C. NSRDS-NBS 13 NSRDS 11 021 46250 ™SRDS.NB^ QC100 -U573 V13;1968 C.1 sH *- NBS-PUB-C 1964 ^f#Cf DftU NBS 'USUCATfONS Hydrogenation of Ethylene Metallic Catalysts U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS National Standard Reference Data Series National Bureau of Standards National Standard Reference Data System, Plan of Operation, NSRDS-NBS 1 — 15 cents* Thermal Properties of Aqueous Uni-univalent Electrolytes NSRDS-NBS 2 — 45 cents* Selected Tables of Atomic Spectra, Atomic Energy Levels and Multiplet Tables — Si II, Si ill, Si iv, NSRDS-NBS 3, Section 1—35 cents* Selected Tables of Atomic Spectra, Atomic Energy Levels and Multiplet Tables — Si I NSRDS — NBS 3, Section 2 — 20 cents* Atomic Transition Probabilities, Volume I, Hydrogen Through Neon, NSRDS-NBS 4 — $2.50* The Band Spectrum of Carbon Monoxide, NSRDS-NBS 5 — 70 cents* Tables of Molecular Vibrational Frequencies. Part 1, NSRDS-NBS 6 — 40 cents* High Temperature Properties and Decomposition of Inorganic Salts. Part 1. Sulfates, NSRDS-NBS 7-35 cents* Thermal Conductivity of Selected Materials, NSRDS-NBS 8 — $1.00* Tables of Biomolecular Gas Reactions, NSRDS-NBS 9 — $2.00* Selected Values of Electric Dipole Moments for Molecules in the Gas Phase, NSRDS- NBS 10 — 40 cents* Tables of Molecular Vibrational Frequencies, Part 2, NSRDS-NBS 11 — 30 cents* Tables for the Rigid Asymmetric Rotor: Transformation Coefficient from Symmetric to Asymmetric Bases Expectation Values of P\ and 4 NSRDS-NBS 12 — in press.