Quantum Chemistry Many-Body Methods on Gpus and Multicore Cpus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Light-Matter Interaction and Optical Spectroscopy from Infrared to X-Rays
24th ETSF Workshop on Electronic Excitations Light-Matter Interaction and Optical Spectroscopy from Infrared to X-Rays Jena, Germany 16 – 20 September 2019 Welcome The workshop series of the European Theoretical Spectroscopy Facility (ETSF) provides a fo- rum for excited states and spectroscopy in condensed-matter physics, chemistry, nanoscience, materials science, and molecular physics attracting theoreticians, code developers, and experi- mentalists alike. Light-matter interaction will be at core of the 2019 edition of the ETSF workshop. Cutting-edge spectroscopy experiments allow to probe electrons, plasmons, excitons, and phonons across different energy and time scales with unprecedented accuracy. A deep physical understanding of the underlying quantum many-body effects is of paramount importance to analyze exper- imental observations and render theoretical simulations predictive. The workshop aims at discussing the most recent advances in the theoretical description of the interaction between light and matter focusing on first-principles methods. This broad subject will be covered in its diverse declinations, from core-level spectroscopy to collective low-energy excitations, dis- cussing also matter under extreme conditions, and systems driven out of equilibrium by strong laser pulses. Exchange between theorists and experimentalists is fostered to open new horizons towards the next generation of novel spectroscopy techniques. The workshop will also face the challenges posed by the formidable complexity of heterogeneous and nanostructured systems such as those of interest for light harvesting and energy generation, prompting to bridge the gap be- tween experimental and in silico spectroscopy. Workshop topics include: Linear and nonlinear optical spectroscopy • Core-level spectroscopies • Ultrafast excitation dynamics • Electron-phonon coupling • Light harvesting in natural and synthetic systems • We are glad to welcome you in Jena, the city of light, and wish you an inspiring workshop with lots of interesting science and fruitful discussions. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

Impact of the Electronic Band Structure in High-Harmonic Generation Spectra of Solids
Impact of the Electronic Band Structure in High-Harmonic Generation Spectra of Solids The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Tancogne-Dejean, Nicolas et al. “Impact of the Electronic Band Structure in High-Harmonic Generation Spectra of Solids.” Physical Review Letters 118.8 (2017): n. pag. © 2017 American Physical Society As Published http://dx.doi.org/10.1103/PhysRevLett.118.087403 Publisher American Physical Society Version Final published version Citable link http://hdl.handle.net/1721.1/107908 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. week ending PRL 118, 087403 (2017) PHYSICAL REVIEW LETTERS 24 FEBRUARY 2017 Impact of the Electronic Band Structure in High-Harmonic Generation Spectra of Solids † Nicolas Tancogne-Dejean,1,2,* Oliver D. Mücke,3,4 Franz X. Kärtner,3,4,5,6 and Angel Rubio1,2,3,5, 1Max Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, 22761 Hamburg, Germany 2European Theoretical Spectroscopy Facility (ETSF), Luruper Chaussee 149, 22761 Hamburg, Germany 3Center for Free-Electron Laser Science CFEL, Deutsches Elektronen-Synchrotron DESY, Notkestraße 85, 22607 Hamburg, Germany 4The Hamburg Center for Ultrafast Imaging, Luruper Chaussee 149, 22761 Hamburg, Germany 5Physics Department, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany 6Research Laboratory of Electronics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA (Received 29 September 2016; published 24 February 2017) An accurate analytic model describing the microscopic mechanism of high-harmonic generation (HHG) in solids is derived. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Popular GPU-Accelerated Applications
LIFE & MATERIALS SCIENCES GPU-ACCELERATED APPLICATIONS | CATALOG | AUG 12 LIFE & MATERIALS SCIENCES APPLICATIONS CATALOG Application Description Supported Features Expected Multi-GPU Release Status Speed Up* Support Bioinformatics BarraCUDA Sequence mapping software Alignment of short sequencing 6-10x Yes Available now reads Version 0.6.2 CUDASW++ Open source software for Smith-Waterman Parallel search of Smith- 10-50x Yes Available now protein database searches on GPUs Waterman database Version 2.0.8 CUSHAW Parallelized short read aligner Parallel, accurate long read 10x Yes Available now aligner - gapped alignments to Version 1.0.40 large genomes CATALOG GPU-BLAST Local search with fast k-tuple heuristic Protein alignment according to 3-4x Single Only Available now blastp, multi cpu threads Version 2.2.26 GPU-HMMER Parallelized local and global search with Parallel local and global search 60-100x Yes Available now profile Hidden Markov models of Hidden Markov Models Version 2.3.2 mCUDA-MEME Ultrafast scalable motif discovery algorithm Scalable motif discovery 4-10x Yes Available now based on MEME algorithm based on MEME Version 3.0.12 MUMmerGPU A high-throughput DNA sequence alignment Aligns multiple query sequences 3-10x Yes Available now LIFE & MATERIALS& LIFE SCIENCES APPLICATIONS program against reference sequence in Version 2 parallel SeqNFind A GPU Accelerated Sequence Analysis Toolset HW & SW for reference 400x Yes Available now assembly, blast, SW, HMM, de novo assembly UGENE Opensource Smith-Waterman for SSE/CUDA, Fast short -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -

Programming Models for Quantum Many-Body Methods on Multicore and Manycore Processors
Programming models for quantum many-body methods on multicore and manycore processors Jeff Hammond1 and Eugene DePrince2 1 Argonne 2 Georgia Tech 6 February 2011 Jeff Hammond Electronic Structure Calculation Methods on Accelerators Abstract The growing diversity in computer processor architectures poses a serious challenge to the computational chemistry community. This talk considers some of the key issues, including disjoint address spaces, non-standard architectures and execution models, and the different APIs required to use them. Specifically, we will describe our experiences in developing coupled-cluster methods for Intel multicore, Intel MIC, NVIDIA Fermi and Blue Gene/Q in both a clean-sheet implementation and NWChem. Of fundamental interest is the development of codes that scale not only within the node but across thousands of nodes; hence, the interaction between the processor and the network will be analyzed in detail. Jeff Hammond Electronic Structure Calculation Methods on Accelerators Software Automation Jeff Hammond Electronic Structure Calculation Methods on Accelerators The practical TCE { NWChem many-body codes What does it do? 1 GUI input quantum many-body theory e.g. CCSD. 2 Operator specification of theory. 3 Apply Wick's theory to transform operator expressions into array expressions. 4 Transform input array expression to operation tree using many types of optimization. 5 Produce Fortran+Global Arrays+NXTVAL implementation. Developer can intercept at various stages to modify theory, algorithm or implementation. Jeff Hammond Electronic Structure Calculation Methods on Accelerators The practical TCE { Success stories First parallel implementation of many (most) CC methods. First truly generic CC code (not string-based): fRHF,ROHF,UHF}×CCfSD,SDT,SDTQ}×{T /Λ,EOM,LR/QRg Most of the largest calculations of their kind employ TCE: CR-EOMCCSD(T), CCSD-LR α, CCSD-QR β, CCSDT-LR α Reduces implementation time for new methods from years to hours, TCE codes are easy to verify. -
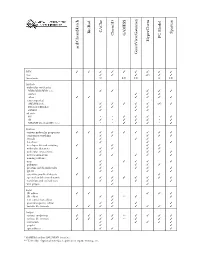
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Propriétés Structurales, Opto-Électroniques, Élastiques Et Dynamiques Des Semi-Conducteurs Type II-VI
REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET POPULAIRE MINISTERE DE L’ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE UNIVERSITE FERHAT ABBAS – SETIF THESE Présentée à la faculté des Sciences Département de Physique Pour l’obtention du diplôme de DOCTORAT EN SCIENCES Option : physique du solide Par Mr. Benamrani Ammar Thème Propriétés structurales, opto-électroniques, élastiques et dynamiques des Semi-conducteurs type II-VI Soutenue le : 02/06/2012 Devant la commission d’examen : Dr. L. Louail Prof. U. F. A. SETIF Président Dr. K. Kassali Prof. U. F. A. SETIF Rapporteur Dr. Kh. Bouamama Prof. U. F. A. SETIF Co-Encadreur Dr. B. Bennecer Prof. U. Guelma Examinateur Dr. A. El Akrmi Prof. U. Annaba Examinateur Dr. H. Belkhir Prof. U. Annaba Examinateur À la mémoire de mon père, À ma mère, qui m’a enseigné le sens de la patience et de l’espoir, À ma femme, À toute ma famille À toute personne qui explore honnêtement les secrets de ce monde vaste, Je dédie ce modeste travail Remerciements REMERCIEMENTS Ce travail a été réalisé au laboratoire d’optoélectronique et des composants à l’université Ferhat Abbes à Sétif. Je tiens à remercier profondément Pr. Kamel Kassali, mon directeur de thèse pour m’avoir proposé ce thème intéressant et pour sa patience durant toutes les années de préparation de la thèse ainsi que ses conseils précieux et ses réponses à toutes mes questions reliées à mon sujet de thèse. J’exprime également toute ma reconnaissance à Mr. Kh. Bouamama, Professeur à l’université Ferhat Abbes de Sétif, qui m’a grandement fait profiter de ses fructueuses discussions en codes de calcul et dans mon sujet de thèse. -

DFT-Absorption Spectra
2054-9 Structure and Dynamics of Hydrogen-Bonded Systems 26 - 27 October 2009 Effect of Proton Disorder on the Excited State Properties of Ice Olivia PULCI Universita' di Roma II "Tor Vergata" Dipt. di Fisica, Via della Ricerca Scientfica 1, 00133 Rome Italy EffectEffect ofof protonproton disorderdisorder onon thethe excitedexcited statestate propertiesproperties ofof iceice V. Garbuio,Garbuio M. Cascella, R. Del Sole, O. Pulci OUTLINE: •Theoretical approaches •Ice Ic(bulk) • Ice Ih surface Theoretical approaches c MBPT c c EXC W hν hν ωcv hν ωcv ωcv v v v DFT GW BSE 1) 2) 3) Ground state properties Electronic band Optical spectra structure, I, A TDDFT (Step 2) Lars Hedin 1965 Σ = iGW G: single particle Green’s function −1 W: screened Coulomb interaction = ε VW Theoretical approaches c MBPT c c EXC W hν hν ωcv hν ωcv ωcv v v v DFT GW BSE 1) 2) 3) Ground state properties Electronic band Optical spectra structure, I, A TDDFT Step 3: calculation of optical spectra within the Bethe Salpeter Equation c Absorption spectra A photon excites an electron from an occupied state to a conduction state hν e 4 4 4 4 4 PPPP=IQP +IQP Ξ v h Bethe Salpeter Equation (BSE) Kernel: Ξ v= − W e-h exchange bound excitons GWBSE ApplicableApplicable to:to: Ab-initio: (NOT “one puts nothing in, one gets nothing out”!!) •Generality, transferability 0D-3D Biological systems •Detailed physical informations •Complex theory+large comp.cost 3-D 0-D 1-D 2-D Nanowires Surfaces Nanoclusters bulks OUTLINE: •Theoretical approaches •Ic Ice (bulk) • Ice Ih surface HH2OO phasephase diagramdiagram CubicCubic iceice ((IcIc)) • It is a metastable form of ice that can be formed, by condensation of water vapor, at ambient pressure but low temperatures Cubic ice (Ic) – diamond lattice 153 K down to 113 K Studied within DFT and Tight-binding G. -

Benchmarking and Application of Density Functional Methods In
BENCHMARKING AND APPLICATION OF DENSITY FUNCTIONAL METHODS IN COMPUTATIONAL CHEMISTRY by BRIAN N. PAPAS (Under Direction the of Henry F. Schaefer III) ABSTRACT Density Functional methods were applied to systems of chemical interest. First, the effects of integration grid quadrature choice upon energy precision were documented. This was done through application of DFT theory as implemented in five standard computational chemistry programs to a subset of the G2/97 test set of molecules. Subsequently, the neutral hydrogen-loss radicals of naphthalene, anthracene, tetracene, and pentacene and their anions where characterized using five standard DFT treatments. The global and local electron affinities were computed for the twelve radicals. The results for the 1- naphthalenyl and 2-naphthalenyl radicals were compared to experiment, and it was found that B3LYP appears to be the most reliable functional for this type of system. For the larger systems the predicted site specific adiabatic electron affinities of the radicals are 1.51 eV (1-anthracenyl), 1.46 eV (2-anthracenyl), 1.68 eV (9-anthracenyl); 1.61 eV (1-tetracenyl), 1.56 eV (2-tetracenyl), 1.82 eV (12-tetracenyl); 1.93 eV (14-pentacenyl), 2.01 eV (13-pentacenyl), 1.68 eV (1-pentacenyl), and 1.63 eV (2-pentacenyl). The global minimum for each radical does not have the same hydrogen removed as the global minimum for the analogous anion. With this in mind, the global (or most preferred site) adiabatic electron affinities are 1.37 eV (naphthalenyl), 1.64 eV (anthracenyl), 1.81 eV (tetracenyl), and 1.97 eV (pentacenyl). In later work, ten (scandium through zinc) homonuclear transition metal trimers were studied using one DFT 2 functional. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11