Chapter 9 Alkynes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
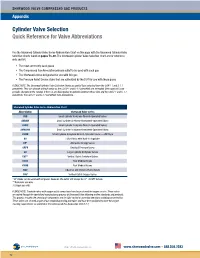
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

There Are Three Major Biological Molecules Classified As Ketone Bodies
There are three major biological molecules classified as ketone bodies: These ketone bodies are water soluble and do not need specific transporters to cross membranes. Synthesis of acetoacetate 1. React two acetyl-CoA molecules with each other using thiolase. This is called acetoacetyl-CoA. What is the second product? 2. React a third acetyl-CoA molecule with acetoacetyl-CoA. This step is catalyzed by hydroxymethylglutaryl-CoA synthase (HMG-CoA synthase). a. Deprotonate C2 of acetyl-CoA. You have created a great nucleophile. b. React your newly formed carbanion nucleophile with the electrophilic carbonyl C3 of acetoacetyl-CoA. c. Protonate the oxyanion. d. Use water as a nucleophile to react with the electrophilic carbonyl of the thioester of the newly added acetyl-CoA unit. This results in a carboxylate functional group. e. Your product should contain a 5-carbon chain, which starts with a thioester to CoA, ends with a carboxylate, and has a hydroxyl and a methyl group attached to C3. This is β-hydroxy-β- methylglutaryl-CoA (HMG-CoA). 3. An acetyl-CoA group is eliminated. This step is catalyzed by hydroxymethylglutaryl-CoA lyase (HMG- CoA lyase). a. A base deprotonates the hydroxyl group of β-hydroxy-β-methylglutaryl-CoA. b. A pair of electrons from the oxyanion moves to form a carbonyl. C2 leaves as a carbanion [which delocalizes into the adjacent thioester carbonyl]. c. The first product is acetoacetate. d. The carbanion picks up the proton and leaves as acetyl-CoA. Formation of acetone from acetoacetate This occurs in a non-enzymatic fashion because of the arrangement of the ketone in the β position from the carboxylate in acetoacetate and causes problems since acetone builds up. -

DEVELOPMENT of NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS by LINDSEY RAE CULLEN DISSERTATION Submitted
DEVELOPMENT OF NOVEL METHODOLOGIES UTILIZING QUATERNARY AMMONIUM SALTS AS CATALYSTS BY LINDSEY RAE CULLEN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Professor Scott E. Denmark, Chair Professor Martin D. Burke Professor Scott K. Silverman Professor Steven C. Zimmerman ii Abstract The first half of this thesis (Chapter 2) described the development of a fluoride-promoted conjugate addition of sulfur-stabilized carbanion nucleophiles to α,β-unsaturated ketones and esters. This reaction was achieved using a substoichiometric amount of TBAF, resulting in high yields on the desired 1,4-addition product. The addition of 1,3-dithianes was given particular focus as a novel method for the preparation of differentially protect 1,4-dicarbonyl compounds. Observation by 13C NMR spectroscopy provided evidence that the reaction proceeds through an ion pair, and attempts to extend this reaction to asymmetric additions using a chiral counterion are presented in detail. The second half of this thesis (Chapter 3) details development of a phase transfer catalyzed [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds. Initial investigation focused on identifying viable substrate classes that would undergo selective [2,3]-rearrangement under phase transfer conditions. Under certain conditions, the [2,3]-sigmatropic rearrangement of allyloxy carbonyl compounds takes place in the presence of a phase transfer agent, providing a rare example of a phase transfer catalyzed unimolecular reaction. In the course of this investigation it was found that catalysis is dependent on several variables including base concentration, catalyst structure, and substrate lipophilicity. -

High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA)
energies Article High Purity/Recovery Separation of Propylene from Propyne Using Anion Pillared Metal-Organic Framework: Application of Vacuum Swing Adsorption (VSA) Majeda Khraisheh 1, Fares AlMomani 1,* and Gavin Walker 2 1 Department of Chemical Engineering, College of Engineering, Qatar University, Doha P.O. Box 2713, Qatar; [email protected] 2 Department of Chemical Sciences, SSPC, Bernal Institute, University of Limerick, V94 T9PX Limerick, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +974-4403-4140 Abstract: Propylene is one of the world’s most important basic olefin raw material used in the produc- tion of a vast array of polymers and other chemicals. The need for high purity grade of propylene is essential and traditionally achieved by the very energy-intensive cryogenic separation. In this study, 2− a pillared inorganic anion SIF6 was used as a highly selective C3H4 due to the square grid pyrazine- based structure. Single gas adsorption revealed a very high C3H4 uptake value (3.32, 3.12, 2.97 and 2.43 mmol·g−1 at 300, 320, 340 and 360 K, respectively). The values for propylene for the same tempera- tures were 2.73, 2.64, 2.31 and 1.84 mmol·g−1, respectively. Experimental results were obtained for the two gases fitted using Langmuir and Toth models. The former had a varied degree of representation of the system with a better presentation of the adsorption of the propylene compared to the propyne system. The Toth model regression offered a better fit of the experimental data over the entire range of pressures. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

(12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi Et Al
USOO8198491 B2 (12) United States Patent (10) Patent No.: US 8,198.491 B2 Masatoshi et al. (45) Date of Patent: Jun. 12, 2012 (54) PROCESS FOR PREPARING 5,986,151 A 1 1/1999 Van Der Puy 2,3,3,3-TETRAFLUOROPROPENE AND $425, 23.99 Esthet al. 1,3,3,3-TETRAFLUOROPROPENE 7,833.434 B2 11/2010 Rao et al. 2005/0245773 Al 1 1/2005 Mukhopadhyay et al. (75) Inventors: Nose Masatoshi, Settsu (JP): Komatsu 2006/0258891 A1 1 1/2006 Mukhopadhyay et al. Yuzo, Settsu (JP); Sugiyama Akinari, 3.s! .k A. 239. SgO et stal al. Situ (JP); Shibanuma Takashi, Settsu 2009,0264689 A1 10, 2009 Rao et al. (JP) 2010/0200798 A1 8, 2010 Rao et al. (73) Assignee: Daikin Industries, Ltd., Osaka (JP) FOREIGN PATENT DOCUMENTS EP O 974 571 1, 2000 (*) Notice: Subject to any disclaimer, the term of this JP 63-211245 9, 1988 patent is extended or adjusted under 35 JP 11-14.0002 5, 1999 U.S.C. 154(b) by 0 days. JP 2007-320896 12/2007 WO 2008/OO2499 1, 2008 WO 2008/OO2500 1, 2008 (21) Appl. No.: 13/057,525 WO 2008/030440 3, 2008 (22) PCT Filed: Jul. 21, 2009 OTHER PUBLICATIONS International Search Report issued Mar. 3, 2010 in International (86). PCT No.: PCT/UP2O09/063314 (PCT) Application No. PCT/JP2009/063314. S371 (c)(1) PCT Written Opinion of the International Searching Authority issued (2), (4) Date:s Feb. 4, 2011 Mar.sia. 3, 2010 in International ((PCT) ) Application NoNo. PCT/JP2009/ Haszeldine et al., “Addition of FreeRadicals to Unsaturated Systems. -

Alkylation of Aromatic Hydrocarbons with Divinylbenzene by Solid Polymeric Oxo Acids
Polymer Journal, Vol. 13, No.9, pp 915-917 (1981) NOTE Alkylation of Aromatic Hydrocarbons with Divinylbenzene by Solid Polymeric Oxo Acids Hiroshi HASEGAWA and Toshinobu HIGASHIMURA Department of Polymer Chemistry, Kyoto University, Kyoto 606, Japan. (Received February 28, 1981) KEY WORDS Solid Polymeric Oxo Acid I Alkylation I Bis(l-arylethyl)- benzene I Nafion-H I Amberlyst 151 Divinylbenzene I Aromatic Hydrocar bons I In our recent papers on the alkylation of aromatic hydrocarbons with styrene (eq 1),u we have shown that solid polymeric oxo acids as catalysts can bring about higher yields of 1-phenyl-1-arylethane (I) than the corresponding soluble oxo acids [e.g., poly(styrenesulfonic acid) (Amberlyst 15) vs. p CH3C6H4S03H; perfluorinated resinsulfonic acid (Nafion-H) vs. CF S0 H]. In view of the easy 3 3 yH3 yH3 separation of the product from the catalyst, the use (2) of these effective polymeric oxo acids should be H H industrially more advantageous than homogeneous (II) processes using soluble acidic catalysts. H H EXPERIMENTAL ) CH3-t-Ar + ift' CH2© ) Aromatic Hydrocarbons © Materials (I) (1) The DVB used in most experiments was an isomeric mixture (m-DVB/p-DVB = 70/30, purity;::: This article deals with the alkylation of aromatic 98%) separated from a commercial mixture of hydrocarbons (toluene and m-xylene) with divinyl DVB (60%) and ethylvinylbenzene (40%) by benzene (DVB) catalyzed by solid polymeric oxo preparative liquid chromatography.5 p-DVB and m acids (Nafion-H and Amberlyst 15). If the two vinyl DVB were isolated from the commercial mixture by groups in DVB, as in the case of styrene, react the method of Storey et a/. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

Identification of a Simplest Hypervalent Hydrogen Fluoride
www.nature.com/scientificreports OPEN Identification of a Simplest Hypervalent Hydrogen Fluoride Anion in Solid Argon Received: 19 December 2016 Meng-Chen Liu1, Hui-Fen Chen2, Chih-Hao Chin1, Tzu-Ping Huang1, Yu-Jung Chen3 & Yu-Jong Accepted: 18 April 2017 Wu1,4 Published: xx xx xxxx Hypervalent molecules are one of the exceptions to the octet rule. Bonding in most hypervalent molecules is well rationalized by the Rundle–Pimentel model (three-center four-electron bond), and high ionic bonding between the ligands and the central atom is essential for stabilizing hypervalent molecules. Here, we produced one of the simplest hypervalent anions, HF−, which is known to deviate from the Rundle–Pimentel model, and identified its ro-vibrational features. High-levelab inito calculations reveal that its bond dissociation energy is comparable to that of dihalides, as supported by secondary photolysis experiments with irradiation at various wavelengths. The charge distribution analysis suggested that the F atom of HF− is negative and hypervalent and the bonding is more covalent than ionic. The octet rule indicates that atoms of the main group elements tend to gain or lose electrons in order to have eight electrons in their valence shells1, 2, similar to the electronic configuration of the noble gases. Therefore, to attain this fully filled electronic configuration, atoms combine to form molecules by sharing their valence electrons to form chemical bonds. This rule is especially applicable to the period 2 and 3 elements. Most molecules are formed by following this rule and the bonding structure of molecules can be easily recognized by using Lewis electron dot diagrams.