A Rare Case of CHILD Syndrome in a Boy Reappears
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Experiences of Rare Diseases: an Insight from Patients and Families
Experiences of Rare Diseases: An Insight from Patients and Families Unit 4D, Leroy House 436 Essex Road London N1 3QP tel: 02077043141 fax: 02073591447 [email protected] www.raredisease.org.uk By Lauren Limb, Stephen Nutt and Alev Sen - December 2010 Web and press design www.raredisease.org.uk WordsAndPeople.com About Rare Disease UK Rare Disease UK (RDUK) is the national alliance for people with rare diseases and all who support them. Our membership is open to all and includes patient organisations, clinicians, researchers, academics, industry and individuals with an interest in rare diseases. RDUK was established by Genetic RDUK is campaigning for a Alliance UK, the national charity strategy for integrated service of over 130 patient organisations delivery for rare diseases. This supporting all those affected by would coordinate: genetic conditions, in conjunction with other key stakeholders | Research in November 2008 following the European Commission’s | Prevention and diagnosis Communication on Rare Diseases: | Treatment and care Europe’s Challenges. | Information Subsequently RDUK successfully | Commissioning and planning campaigned for the adoption of the Council of the European into one cohesive strategy for all Union’s Recommendation on patients affected by rare disease in an action in the field of rare the UK. As well as securing better diseases. The Recommendation outcomes for patients, a strategy was adopted unanimously by each would enable the most effective Member State of the EU (including use of NHS resources. the -

Opsoclonus-Myoclonus Syndrome
OMS Opsoclonus-Myoclonus Syndrome REGISTRY POWERED BY NORD 10 11 Tr io Health © 2019 Trio Health Advisory Group, Inc.; NORD - National Organization for Rare Disorders, Inc. | All rights reserved. © 2019 Trio Health Advisory Group, Inc.; NORD - National Organization for Rare Disorders, Inc. | All rights reserved. Tr io Health Meet OMS Warrior ALEXA What is OMS? OPSOCLONUS-MYOCLONUS SYNDROME General Discussion Opsoclonus-myoclonus syndrome (OMS) is an inflammatory neurological disorder, often occurring as a paraneoplastic syndrome with neurological symptoms being the first sign of an occult tumor. It is characterized by associated ocular, motor, behavioral, sleep, and language disturbances. The onset is oftentimes abrupt and can be relatively severe, with the potential to become chronic unless the appropriate diagnosis and treatment are reached in a timely manner. Signs and Symptoms The component features of OMS include the presence of rapid, seemingly random eye movements in the horizontal, vertical, and diagonal directions (opsoclonus); an unsteady gait or inability to walk or stand (ataxia); and brief, repetitive, shock-like muscle spasms or tremors within the arms, legs, or hands interfering with normal use (myoclonus). Behavioral and sleep disturbances, including extreme irritability, inconsolable crying, reduced or fragmented sleep (insomnia), and rage attacks are common. Difficulty articulating speech (dysarthria), sometimes with complete loss of speech and language, may occur. Additional symptoms, such as decreased muscle tone (hypotonia) and vomiting, have also been noted. Causes The most common cause of OMS in young children is an occult (ie, a small, often hidden) tumor. The symptoms of OMS, as a paraneoplastic syndrome, presumably stem from the immune system attacking the tumor, leading to secondary inflammatory effects on the central nervous system. -

Psykisk Utviklingshemming Og Forsinket Utvikling
Psykisk utviklingshemming og forsinket utvikling Genpanel, versjon v03 Tabellen er sortert på gennavn (HGNC gensymbol) Navn på gen er iht. HGNC >x10 Andel av genet som har blitt lest med tilfredstillende kvalitet flere enn 10 ganger under sekvensering x10 er forventet dekning; faktisk dekning vil variere. Gen Gen (HGNC Transkript >10x Fenotype (symbol) ID) AAAS 13666 NM_015665.5 100% Achalasia-addisonianism-alacrimia syndrome OMIM AARS 20 NM_001605.2 100% Charcot-Marie-Tooth disease, axonal, type 2N OMIM Epileptic encephalopathy, early infantile, 29 OMIM AASS 17366 NM_005763.3 100% Hyperlysinemia OMIM Saccharopinuria OMIM ABCB11 42 NM_003742.2 100% Cholestasis, benign recurrent intrahepatic, 2 OMIM Cholestasis, progressive familial intrahepatic 2 OMIM ABCB7 48 NM_004299.5 100% Anemia, sideroblastic, with ataxia OMIM ABCC6 57 NM_001171.5 93% Arterial calcification, generalized, of infancy, 2 OMIM Pseudoxanthoma elasticum OMIM Pseudoxanthoma elasticum, forme fruste OMIM ABCC9 60 NM_005691.3 100% Hypertrichotic osteochondrodysplasia OMIM ABCD1 61 NM_000033.3 77% Adrenoleukodystrophy OMIM Adrenomyeloneuropathy, adult OMIM ABCD4 68 NM_005050.3 100% Methylmalonic aciduria and homocystinuria, cblJ type OMIM ABHD5 21396 NM_016006.4 100% Chanarin-Dorfman syndrome OMIM ACAD9 21497 NM_014049.4 99% Mitochondrial complex I deficiency due to ACAD9 deficiency OMIM ACADM 89 NM_000016.5 100% Acyl-CoA dehydrogenase, medium chain, deficiency of OMIM ACADS 90 NM_000017.3 100% Acyl-CoA dehydrogenase, short-chain, deficiency of OMIM ACADVL 92 NM_000018.3 100% VLCAD -

Tests Performed Through Our International Collaboration
Tests performed through our international collaboration Molecular Studies for Inborn Errors of Metabolism A Inborn Errors of Metabolism Defective Gene 1 Acyl - Co A oxidase deficiency ACOX 1 2 Argininosuccinate lyase deficiency ASL 3 Aromatic L – amino acid decarboxylase (AADC) DDC Deficiency 4 Carnitine – acylcarnitine translocase (CACT) CACT Deficiency 5 Primary (systemic) carnitine deficiency OCTN2 6 Carnitine palmitoyltransferase I (CPT1) CPT1A 7 Carnitine palmitoyltransferase 2 (CPT2) CPT2 8 CHILD Syndrome NSDHL 9 Conradi – Hunermann – Happle syndrome EBP (CDPX2) 10 D – Bifunctional protein (DBP) Deficiency DBP, MFE2 11 Desmosterolosis DHCR24 12 Dihydropyrimidinase (DHP) Deficiency DPYS 13 Dihydropyrimidine dehydrogenase (DPD) DPYD Deficiency 14 Ethylmalonaciduria ETHE1 (Ethylmalonic encephalopathy) 15 Fructose intolerance, hereditary ALDOB 16 Galactosemia, classic GALT 17 Galactokinase deficiency GALK1 18 Glutaric aciduria type I (Glutaryl – Co A GCDH dehydrogenase deficiency 19 Glycogen storage disease 0 GYS2 20 Greenberg skeletal dysplasia LBR (Sterol – delta 14 reductase deficiency) 21 GTP cyclohydrolase I deficiency GCH1 22 Hydroxyacyl – Co A dehydrogenase deficiency HADH2 (2 – Methyl – 3 – hydroxybutyryl – CoA dehydrogenase deficiency) 23 Hyper Ig D Syndrome MVK (Mevalonate Kinase deficiency) 24 Hyperoxaluria type I AGXT 25 Isovaleric acidemia IVD 26 Lathosterolosis SC5DL 27 3 – methylglutaconicaciduria type I (3 – AUH methylglutaconyl – CoA hydratase deficiency) 28 Medium – chain – acyl – Co A dehydrogenase ACADM (MCAD) Deficiency -

Maternal and Child Health Thesaurus Third Edition
Maternal and Child Health Thesaurus Third Edition Compiled by: Olivia K. Pickett, M.A., M.L.S. Director of Library Services Maternal and Child Health Library National Center for Education in Maternal and Child Health Georgetown University Published by: Maternal and Child Health Library National Center for Education in Maternal and Child Health Georgetown University 2115 Wisconsin Avenue, N.W., Suite 601 Washington, DC 20007-2292 (202) 784-9770 [email protected] © 2005 by National Center for Education in Maternal and Child Health and Georgetown University. TABLE OF CONTENTS Introduction ...........................................................................................................................i Alphabetical List................................................................................................................... 3 Rotated Term List............................................................................................................. 147 Subject Categories ............................................................................................................ 213 MCH Thesaurus Introduction INTRODUCTION This third edition of the Maternal and Child Health Thesaurus was developed by the Maternal and Child Health Library, National Center for Education in Maternal and Child Health (NCEMCH), at Georgetown University, under Cooperative Agreement U02MC00001 with the Maternal and Child Health Bureau (MCHB), Health Resources and Services Administration, U.S. Department of Health and Human Services. The Maternal -

Genetic Testing Stories
genetic alliance monograph series number 2 Genetic testinG stories nicole exe, ms heather Ferguson, ms, cgc alyson Krokosky samantha sawyer sharon F. terry, ma published by genetic alliance Genetic testinG stories nicole exe, ms heather Ferguson, ms, cgc alyson Krokosky samantha sawyer sharon F. terry, ma published by genetic alliance 1 Genetic Alliance is grateful to the individuals and families who so generously shared their stories. They offer us a model of openness that benefits all. Partial funding for this report was provided by the Genetics and Public Policy Center. The Center is supported at Johns Hopkins University by The Pew Charitable Trusts. Any opinions expressed in this report are those of the author(s) and do not necessarily reflect the views of The Pew Chari- table Trusts or the Genetics and Public Policy Center. This report may not be reproduced or distributed in whole or in part without the permission of the Genetics and Public Policy Center. Funding for this work was also provided by the Health Resources and Services Administration, Maternal Child Health Bureau, Genetics Services Branch, cooperative agreement # 1 U33MC07945-01-00, and Genetic Alliance. 4301 connecticut ave., nW, suite 404 Washington, dc 20008 t: 202.966.5557 F: 202.966.8553 www.geneticalliance.org 2 Table of Contents PREFACE 4 HYPOKALEMIC PERIODIC PARALYSIS Adult 26 CARRIER TESTING 5 HYPOSPADIAS/CHROMOSOMAL DEFECTS Child 27 22Q11.2 DELETION SYNDROME KLINEFELTER SYNDROME Adult 27 Adult/Newborn 5 MEDIUM CHAIN ACYL COA DEHYDROGENASE ADRENOLEUKODYSTROPHY -

The American Board of Pediatrics® Content Outline
THE AMERICAN BOARD OF PEDIATRICS® CONTENT OUTLINE General Pediatrics In-Training, Certification, and Maintenance of Certification Examinations Effective for examinations administered beginning January 2016 i INTRODUCTION This document was prepared by the American Board of Pediatrics for the purpose of developing certification examinations for general pediatricians. The outline, which was developed by a committee of pediatric practitioners and educators, contains the categories that will be reflected in the general pediatrics examinations. If you have comments or questions about these content specifications, or about how they are used, please send an e-mail to [email protected]. ii General Pediatrics Exam Percentage List Approximate Percent Page I. Growth and Development .................................................... 5.0 ................................... 1 II. Nutrition and Nutritional Disorders ..................................... 4.0 ................................... 2 III. Preventive Pediatrics ............................................................ 5.0 ................................... 5 IV. Poisoning and Environmental Exposure to Hazardous Substances ................................................. 2.0 ................................... 8 V. Fetus and Newborn Infant .................................................... 3.5 ................................... 9 VI. Fluid and Electrolyte Metabolism ........................................ 2.5 ................................. 12 VII. Genetics and Dysmorphology ............................................. -

Conradi-Hünermann-Happle Syndrome (X-Linked Dominant Chondrodysplasia Punctata) Confirmed by Plasma Sterol and Mutation Analysis
Acta Derm Venereol 2008; 88: 47–51 CLINICAL REPORT Conradi-Hünermann-Happle Syndrome (X-linked Dominant Chondrodysplasia Punctata) Confirmed by Plasma Sterol and Mutation Analysis Annette KOLB-MÄURER1, Karl-Heinz GRZESCHIK2, Dorothea HAAS3, Eva-Bettina BRÖCKER1 and Henning HAMM1 1Department of Dermatology, University of Würzburg, Würzburg, 2Department of Human Genetics, University of Marburg, Marburg, 3University Hospital for Paediatric and Adolescent Medicine, Heidelberg, Germany Conradi-Hünermann-Happle syndrome, or X-linked forms to mild, clinically almost undetectable, mani- dominant chondrodysplasia punctata, is a rare genetic festations. The syndrome occurs almost exclusively in disorder characterized by skeletal dysplasia, stippled females and is characterized by skeletal abnormalities, epiphyses, cataracts, transient ichthyosis and atrophic cataracts and characteristic skin lesions. Cutaneous residua in a mosaic pattern. Mutations in the gene enco- involvement typically starts as a severe congenital icht- ding the emopamil-binding protein have been identified hyosis with adherent large scales following the lines of as an underlying cause. A 5-year-old girl presented for Blaschko on an erythrodermic background. Collodion evaluation of ill-defined patches of cicatricial alopecia. baby may precede this typical appearance. After some In addition, subtle follicular atrophoderma, esotropia, months, the ichthyosiform lesions regress and follicular craniofacial asymmetry and short stature were noted. atrophoderma remains. These patchy or linear, atrophic Her history revealed widespread scaly erythema and lesions with follicular accentuation, often on a hypo- or eye surgery for congenital cataract in the first months hyper-pigmented base, are reminiscent of orange-peel of life. Diagnosis of Conradi-Hünermann-Happle syn- skin (2). Mild ichthyosis may persist, particularly on the drome was confirmed by plasma sterol analysis showing limbs. -
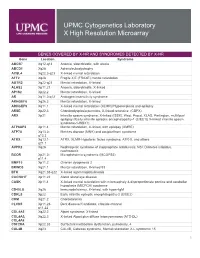
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

The Clinical Spectrum of Congenital
Acta Derm Venereol 2003, Suppl. 213: 34–47 The Clinical Spectrum of Congenital Ichthyosis in Sweden: A Review of 127 Cases ANDERS VAHLQUIST1, AGNETA GÅNEMO1, MARITTA PIGG2, MARIE VIRTANEN1 AND PER WESTERMARK3 Departments of 1Dermatology, 2Clinical Genetics and 3Pathology, University Hospital, Uppsala, Sweden Congenital ichthyosis comprises a rare group of usual- microscopy, HI: Harlequin ichthyosis, IFAP: ichthyosis follicularis, ly monogenetic diseases that present at birth as a collo- alopecia and photofobia, LI-TGM: lamellar ichthyosis with dion phenotype or as variable degrees of ichthyosiform transglutaminase 1 gene mutations, LI/CIE: lamellar ichthyosis and/ or congenital ichthyosiform erythroderma, KID: keratitis, ichthyo- erythroderma, with or without superficial blisters. De- sis and deafness, K: keratin, KLICK: keratosis linearis, ichthyosis pending on which gene mutation causes the disease, the congenita and keratoderma, SLS: Sjögren-Larsson syndrome, Tgase skin problems later in life may range from a severe 1 – transglutaminase 1 protein, XRI: X-linked recessive ichthyosis. lamellar or bullous ichthyosis to mild or only focally expressed hyperkeratotic lesions. It is obviously impor- tant, but sometimes painstakingly difficult, to make a INTRODUCTION correct diagnosis already in infancy. Fortunately, re- cent advances in our understanding of the molecular Congenital ichthyosis (CI) encompasses a large group of genetics of ichthyosis have led to several new diagnos- mostly monogenetic disorders of keratinization present- tic tools that are continuously being updated. Based on ing at birth as widespread hyperkeratosis and scaling of the integument, and sometimes associated with erythro- Downloaded By: [Akademiska Sjukhuset] At: 12:48 5 October 2007 this development, and on our own 5 years of experi- ence in a national genodermatosis centre, we describe derma and skin erosions. -

Peroxisomes and Metabolic Disease
ì'gt PEROXISOMES AND METABOLIC DISEASE JENNIFER LUCY HUGHES Thesis submitted for the degree of Doctor of Philosophy in The University of Adelaide (Faculty of Medicine) A,^,and."\ ìqq + ii dedication t dedícate this work to my husband Gregory John Hanisch, and my children Max (deceased), Nicholas Oscar and Jay Alexander. ¡¡i declaration This work contains no mater¡al which has been accepted for the award of any other degree or diploma in any university or other tertiary institutlon and, to the best of my knowledge and belief, contains no mater¡al previously published or written by another person, except where due reference has been made ¡n the text. I give consent to th¡s copy of my thesis, when deposited in the Un¡versity Library, being available for loan and photocopying. SIGNED: oere:2*:.).:3.:t- iv summary Peroxisomes are cellular organelles which carry out many metabolic functions including in particular the breakdown and synthesis of fats. Peroxisomes are limited by a single membrane, and the integral membrane proteins and the constituent matrix enzymes are synthesized on free polyribosomes and incorporated post-translationally into pre-existing organelles. The mechanisms involved in this biosynthetic process are not fully understood. There is a group of inherited peroxisomal disorders, which are characterised biochemically by a deficiency of one or more peroxisomal enzymes, and an accumulation of metabolites in the ! ¡,r, patient's tissues and plasma. The affected children most often die before reaching one year of age and suffer severe clinical symptoms including abnormalities of the liver, kidney, brain, adrenal and b_o_ne. -

Connective Tissue Disorders
UNIVERSITY OF MINNESOTA PHYSICIANS OUTREACH LABS Submit this form along with the appropriate Molecular requisition (Molecular Diagnostics or MOLECULAR DIAGNOSTICS (612) 273-8445 Molecular NGS Oncology). DATE: TIME COLLECTED: PCU/CLINIC: AM PM PATIENT IDENTIFICATION DIAGNOSIS (Dx) / DIAGNOSIS CODES (ICD-9) - OUTPATIENTS ONLY SPECIMEN TYPE: o Blood (1) (2) (3) (4) PLEASE COLLECT 5-10CC IN ACD-A OR EDTA TUBE ORDERING PHYSICIAN NAME AND PHONE NUMBER: Tests can be ordered as a full panel, or by individual gene(s). Please contact the genetic counselor with any questions at 612-624-8948 or by pager at 612-899-3291. _______________________________________________ Test Ordered- EPIC: Next generation sequencing(Next Gen) Sunquest: NGS FBN1 Connective tissue Asphyxiating thoracic dystrophy Cranioectodermal dysplasia 2 disorders Full panel WDR35 IFT80 Achondrogenesis DYNC2H1 Craniolenticulosutural dysplasia Full panel TTC21B SEC23A SLC26A2 WDR19 Craniosynostosis TRIP11 Atelosteogenesis Full panel COL2A1 Full panel FGFR2 Achondroplasia SLC26A2 IL11RA FGFR3 FLNB CYP26B1 Acrocapitofemoral dysplasia Brachydactyly TWIST1 IHH Full panel MSX2 Acrodysostosis GDF5 FGFR1 Full panel BMP2 FGFR3 PRKAR1A PTHLH PDE4D Brachyolmia Cutis laxa Acrofacial dysostosis Full panel Full panel Full panel PAPSS2 ATP6V0A2 SF3B4 TRPV4 EFEMP2 DHODH Campomelic dysplasia with autosomal PYCR1 Acromesomelic dysplasia sex reversal ALDH18A1 Full panel SOX9 GDF5 Camurati-Engelmann disease De la Chapelle dysplasia NPR2 TGFB1 SLC26A2 Alport syndrome Cenani-Lenz syndactyly syndrome Full