Phosgene Final AEGL Document
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Warning: the Following Lecture Contains Graphic Images
What the новичок (Novichok)? Why Chemical Warfare Agents Are More Relevant Than Ever Matt Sztajnkrycer, MD PHD Professor of Emergency Medicine, Mayo Clinic Medical Toxicologist, Minnesota Poison Control System Medical Director, RFD Chemical Assessment Team @NoobieMatt #ITLS2018 Disclosures In accordance with the Accreditation Council for Continuing Medical Education (ACCME) Standards, the American Nurses Credentialing Center’s Commission (ANCC) and the Commission on Accreditation for Pre-Hospital Continuing Education (CAPCE), states presenters must disclose the existence of significant financial interests in or relationships with manufacturers or commercial products that may have a direct interest in the subject matter of the presentation, and relationships with the commercial supporter of this CME activity. The presenter does not consider that it will influence their presentation. Dr. Sztajnkrycer does not have a significant financial relationship to report. Dr. Sztajnkrycer is on the Editorial Board of International Trauma Life Support. Specific CW Agents Classes of Chemical Agents: The Big 5 The “A” List Pulmonary Agents Phosgene Oxime, Chlorine Vesicants Mustard, Phosgene Blood Agents CN Nerve Agents G, V, Novel, T Incapacitating Agents Thinking Outside the Box - An Abbreviated List Ammonia Fluorine Chlorine Acrylonitrile Hydrogen Sulfide Phosphine Methyl Isocyanate Dibotane Hydrogen Selenide Allyl Alcohol Sulfur Dioxide TDI Acrolein Nitric Acid Arsine Hydrazine Compound 1080/1081 Nitrogen Dioxide Tetramine (TETS) Ethylene Oxide Chlorine Leaks Phosphine Chlorine Common Toxic Industrial Chemical (“TIC”). Why use it in war/terror? Chlorine Density of 3.21 g/L. Heavier than air (1.28 g/L) sinks. Concentrates in low-lying areas. Like basements and underground bunkers. Reacts with water: Hypochlorous acid (HClO) Hydrochloric acid (HCl). -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Nonflammable Gas Mixture: Arsine / Helium / Phosgene Section 1. Identification GHS product identifier : Nonflammable Gas Mixture: Arsine / Helium / Phosgene Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 006605 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Compressed gas substance or mixture ACUTE TOXICITY (inhalation) - Category 4 GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. Harmful if inhaled. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Prevention : Use only outdoors or in a well-ventilated area. Avoid breathing gas. Response : IF INHALED: Remove person to fresh air and keep comfortable for breathing. Call a POISON CENTER or physician if you feel unwell. Storage : Protect from sunlight when ambient temperature exceeds 52°C/125°F. Store in a well- ventilated place. -

OSHA Method 61: Phosgene
PHOSGENE - (Organic Method #61) Page 1 of 20 U.S. Department of Labor Occupational Safety & Health Administration www.osha.gov Search Advanced Search | A Technical Links > Sampling & Analytical Methods > Index PHOSGENE Method no.: 61 Matrix: Air Target concentration: 100 ppb (0.4 mg/m3) (OSHA PEL) Procedure: Air samples are collected by drawing known volumes of air through sampling tubes containing XAD-2 adsorbent which has been coated with 2 (hydroxymethyl)piperidine. The samples are desorbed with toluene and then analyzed by gas chromatography using a nitrogen selective detector. Recommended air volume and sampling rate: 240 L at 1 L/min Reliable quantitation limit: 3.5 ppb (0.014 mg/m3) Standard error of estimate at the target concentration: 6.7% (Section 4.7.) Status of method: Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. Date: August 1986 Chemist: Warren Hendricks Organic Methods Evaluation Branch OSHA Analytical Laboratory Salt Lake City, Utah 1. General Discussion 1.1. Background 1.1.1. History http://www.osha.gov/dts/sltc/methods/organic/org061/org061.html 7/7/2008 PHOSGENE - (Organic Method #61) Page 2 of 20 The procedures that have been used by OSHA to monitor occupational exposure to phosgene include detector tubes, monitoring dosimeters and infrared gas analyzers. None of these procedures have proven to be completely adequate for use by OSHA. These methods lack either the desired precision and accuracy or they are awkward and inconvenient for field use. This procedure was developed after it was found that phosgene would react quantitatively with 2-(hydroxymethyl)piperidine (2-HMP) to form a stable derivative, 1-aza-8-oxabicyclo[4.3.0.]inonan-9-one. -

Inorganic Arsenic Compounds Other Than Arsine Health and Safety Guide
OS INTERNATiONAL I'ROGRAMME ON CHEMICAL SAFETY Health and Safety Guide No. 70 INORGANIC ARSENIC COMPOUNDS OTHER THAN ARSINE HEALTH AND SAFETY GUIDE i - I 04 R. Q) UNEP UNITED NATIONS INTERNATIONAL ENVIRONMENT I'R( )GRAMME LABOUR ORGANISATION k\s' I V WORLD HEALTH ORGANIZATION WORLD HEALTH ORGANIZATION, GENEVA 1992 IPcs Other H EA LTH AND SAFETY GUIDES available: Aerytonitrile 41. Clii rdeon 2. Kekvau 42. Vatiadiuni 3 . I Bula not 43 Di meLhyI ftirmatnide 4 2-Buta101 44 1-Dryliniot 5. 2.4- Diehlorpheiioxv- 45 . Ac rylzi mule acetic Acid (2.4-D) 46. Barium 6. NIcihylene Chhride 47. Airaziiie 7 . ie,i-Buia nol 48. Benlm'.ie 8. Ep Ichioroli) Olin 49. Cap a 64 P. ls.ihutaiiol 50. Captaii I o. feiddin oeth N lene Si. Parai.tuat II. Tetradi ion 51 Diquat 12. Te nacelle 53. Alpha- and Betal-lexachloro- 13 Clils,i (lane cyclohexanes 14 1 kpia Idor 54. Liiidaiic IS. Propylene oxide 55. 1 .2-Diciilroetiiane Ethylene Oxide 5t. Hydrazine Eiulosiillaii 57. F-orivaldehydc IS. Die h lorvos 55. MLhyI Isobu I V I kcloiic IV. Pculaehloro1heiiol 59. fl-Flexaric 20. Diiiiethoaie 61), Endrin 2 1 . A iii in and Dick) 0in 6 I . I sh IIZiLI1 22. Cyperniellirin 62. Nicki. Nickel Caution I. and some 23. Quiiiloieiic Nickel Compounds 24. Alkthrins 03. Hexachlorocyclopeuladiene 25. Rsiiiethii ins 64. Aidicaib 26. Pyr rot ii,id inc Alkaloids 65. Fe nitrolhioit 27. Magnetic Fields hib. Triclilorlon 28. Phosphine 67. Acroleiii 29. Diiiiethyl Sull'ite 68. Polychlurinated hiphenyls (PCBs) and 30. Dc lianteth nil polyc h In ruiated letlilienyls (fs) 31. -

Arsine Final AEGL Document
Acute Exposure Guideline Levels for Selected Airborne Chemicals Volume 1 Subcommittee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Commission of Life Sciences National Research Council NATIONAL ACADEMY PRESS Washington, DC NATIONAL ACADEMY PRESS 2101 Constitution Ave., N.W. Washington, D.C. 20418 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract Nos. DAMD17-89-C-9086 and DAMD17-99-C-9049 between the National Academy of Sciences and the U.S. Army. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number 0-309-07294-8 Additional copies of this report are available from: National Academy Press 2101 Constitution Ave., NW Box 285 Washington, DC 20055 800-624-6242 202-334-3313 (in the Washington metropolitan area) http://www.nap.edu Copyright 2000 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare. -

0163 Date: June 1998 Revision: February 2008 DOT Number: UN 2188
Right to Know Hazardous Substance Fact Sheet Common Name: ARSINE Synonyms: Arsenic Hydride; Hydrogen Arsenide CAS Number: 7784-42-1 Chemical Name: Arsine RTK Substance Number: 0163 Date: June 1998 Revision: February 2008 DOT Number: UN 2188 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE Arsine is a colorless gas with a garlic-like odor. It is used in Hazard Summary making electronic components, in organic synthesis, in making Hazard Rating NJDOH NFPA lead acid storage batteries, and as a military poison gas. HEALTH - 4 FLAMMABILITY - 4 Exposure to Arsine often occurs when Arsine is formed as a REACTIVITY - 2 by-product of a chemical reaction between Arsenic, or a Base CARCINOGEN Metal containing Arsenic impurities, and an acid or strong alkali FLAMMABLE AND REACTIVE (base). POISONOUS GASES ARE PRODUCED IN FIRE CONTAINERS MAY VENT RAPIDLY AND EXPLODE IN FIRE Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; Reasons for Citation 4=severe f Arsine is on the Right to Know Hazardous Substance List because it is cited by OSHA, ACGIH, DOT, NIOSH, NTP, f Arsine can affect you when inhaled. DEP, IARC, IRIS, NFPA and EPA. f Arsine is a CARCINOGEN. HANDLE WITH EXTREME f This chemical is on the Special Health Hazard Substance CAUTION. List. f Skin and eye contact with liquid can cause frostbite. f Inhaling Arsine can irritate the lungs. Higher exposures may cause a build-up of fluid in the lungs (pulmonary edema), a medical emergency. f High exposure to Arsine can destroy red blood cells SEE GLOSSARY ON PAGE 5. (hemolysis), causing anemia. -

Toxic Exposures Kathy L
8 MODULE 8 Toxic Exposures Kathy L. Leham-Huskamp / William J. Keenan / Anthony J. Scalzo / Shan Yin 8 Toxic Exposures Kathy L. Lehman-Huskamp, MD William J. Keenan, MD Anthony J. Scalzo, MD Shan Yin, MD InTrODUcTIOn The first large-scale production of chemical and biological weapons occurred during the 20th century. World War I introduced the use of toxic gases such as chlorine, cyanide, an arsine as a means of chemical warfare. With recent events, such as the airplane attacks on the World Trade Center in New York City, people have become increasingly fearful of potential large-scale terrorist attacks. Consequently, there has been a heightened interest in disaster preparedness especially involving chemical and biological agents. The U.S. Federal Emergency Management Agency (FEMA) recommends an "all-hazards" approach to emergency planning. This means creating a simultaneous plan for intentional terrorist events as well as for the more likely unintentional public health emergencies, such as earthquakes, floods, hazardous chemical spills, and infectious outbreaks. Most large-scale hazardous exposures are determined by the type of major industries that exist and/or the susceptibility to different types of natural disasters in a given area. For example, in 1984 one of the greatest man-made disasters of all times occurred in Bhopal, India, when a Union Carbide pesticide plant released tons of methylisocyanate gas over a populated area, killing scores of thousands and injuring well over 250,000 individuals. The 2011 earthquake and tsunami in Japan demonstrated the vulnerability of nuclear power stations to natural disasters and the need to prepare for possible widespread nuclear contamination and radiation exposure. -

Investigation and Risk Assessment of Ships Loaded with Chemical Ammunition Scuttled in Skagerrak
Investigation and risk assessment of ships loaded with chemical ammunition scuttled in Skagerrak TA-1907/2002 Tørnes John Aa, Voie Øyvind A, Ljønes Marita, Opstad Aase M, Bjerkeseth Leif Haldor, Hussain Fatima Investigation and risk assessment of ships loaded with chemical ammunition scuttled in Skagerrak (TA-1907/2002) Preface The current report gives a description of the investigation carried out by Forsvarets forskningsinstitutt (FFI) of the wrecks dumped in Skagerrak after World War II with chemical ammunition on board. The aim of the investigation was to give an evaluation of the conditions of the dumped wrecks and to assess whether the chemical warfare agents have leaked out from the wrecks. This was done by using a remote-operated vehicle with video cameras. Sediment samples were collected and analysed with respect to chemical warfare agents and some related compounds. Temperature and sea current at the sea bottom was measured. This made it possible to give a rough estimate of the risks associated with leaking ammunition. Some recommendation for further work is also given. FFI, Kjeller, November 2002 Bjørn A Johnsen Director of Research 3 Investigation and risk assessment of ships loaded with chemical ammunition scuttled in Skagerrak (TA-1907/2002) 4 Investigation and risk assessment of ships loaded with chemical ammunition scuttled in Skagerrak (TA-1907/2002) Contents 1. Summary..................................................................................................7 1.1 Background ................................................................................................................. -

Acetylcholinesterase: the “Hub” for Neurodegenerative Diseases And
Review biomolecules Acetylcholinesterase: The “Hub” for NeurodegenerativeReview Diseases and Chemical Weapons Acetylcholinesterase: The “Hub” for Convention Neurodegenerative Diseases and Chemical WeaponsSamir F. de A. Cavalcante Convention 1,2,3,*, Alessandro B. C. Simas 2,*, Marcos C. Barcellos 1, Victor G. M. de Oliveira 1, Roberto B. Sousa 1, Paulo A. de M. Cabral 1 and Kamil Kuča 3,*and Tanos C. C. França 3,4,* Samir F. de A. Cavalcante 1,2,3,* , Alessandro B. C. Simas 2,*, Marcos C. Barcellos 1, Victor1 Institute G. M. ofde Chemical, Oliveira Biological,1, Roberto Radiological B. Sousa and1, Paulo Nuclear A. Defense de M. Cabral (IDQBRN),1, Kamil Brazilian Kuˇca Army3,* and TanosTechnological C. C. França Center3,4,* (CTEx), Avenida das Américas 28705, Rio de Janeiro 23020-470, Brazil; [email protected] (M.C.B.); [email protected] (V.G.M.d.O.); [email protected] 1 Institute of Chemical, Biological, Radiological and Nuclear Defense (IDQBRN), Brazilian Army (R.B.S.); [email protected] (P.A.d.M.C.) Technological Center (CTEx), Avenida das Américas 28705, Rio de Janeiro 23020-470, Brazil; 2 [email protected] Mors Institute of Research (M.C.B.); on Natural [email protected] Products (IPPN), Federal (V.G.M.d.O.); University of Rio de Janeiro (UFRJ), CCS,[email protected] Bloco H, Rio de Janeiro (R.B.S.); 21941-902, [email protected] Brazil (P.A.d.M.C.) 32 DepartmentWalter Mors of Institute Chemistry, of Research Faculty of on Science, Natural Un Productsiversity (IPPN), -

Analysis of Arsine and Phosphine in Ethylene and Propylene Using the Agilent Arsine Phosphine GC/MS Analyzer with a High Effi Ciency Source
Analysis of Arsine and Phosphine in Ethylene and Propylene Using the Agilent Arsine Phosphine GC/MS Analyzer with a High Effi ciency Source Application Note Petrochemical Authors Abstract Angela Smith Henry and Bruce Quimby The Agilent Arsine Phosphine GC/MS analyzer enables detection at single-digit Agilent Technologies Inc. parts per billion (ppb) concentrations and long-term signal stability of arsine, Wilmington, DE phosphine, and other contaminants in ethylene and propylene, with relative standard deviations of ~5 % or less. Introduction Instruments The Agilent Arsine Phosphine GC/MS analyzer (G3440B with Developments in the area of metallocene catalysts have option SP1 789 0-0667) uses the Agilent 7890B GC and the signifi cantly increased productivity for the polymerization of Agilent 5977B Series GC/MSD System HES with hydrogen ethylene and propylene. However, these catalysts can also cleaning (Figure 1). Thick fi lm columns provide resolution of be more susceptible to impurities such as arsine (AsH ), 3 the four contaminants in ethylene and three contaminants phosphine (PH3), hydrogen sulfi de (H2S), and carbonyl sulfi de (PH3, AsH3, and H2S) in propylene. Table 1 lists the analysis (COS). This sensitivity to contaminants has driven a need to conditions. monitor impurities at the lowest possible detection levels. Contaminants can degrade a polymerization catalyst sooner 2-Port valve system than desired, and can potentially shut down the production SCIS process for catalyst replacement. Precise, low-level detection 5 4 3 H2 6 3 4 2 of these contaminants during the production process offers 1 2 1 the ability for olefi n producers to take steps to mitigate these He contaminants. -
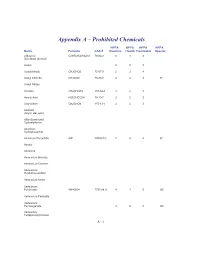
Appendix a – Prohibited Chemicals
Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special 2-Butanol C2H5CH(OH)CH3 78-92-2 0 1 3 (Sec-Butyl Alcohol) Acetal 023 Acetaldehyde CH3CHO5 75-07-0 2 3 4 Acetyl Chloride CH3COCl 75-36-5 2 3 3 W Acetyl Nitrate Acrolein CH2CHCHO 107-02-8 3 4 3 Acrylic Acid H2CCHCO2H 79-10-7 2 2 2 Acrylonitrile CH2CHCN 107-13-1 2 4 3 Alcohols (Allylic, Benzylic) Alkly-Substituted Cycloaliphatics Aluminum Hydrophosphide Aluminum Phosphide AlP 20859-73- 2 4 4 W Amatol Ammonal Ammonium Bromate Ammonium Chlorate Ammonium Hexanitrocobaltate Ammonium Nitrite Ammonium Perchlorate NH4ClO4 7790-98-9 4 1 0 OX Ammonium Periodate Ammonium Permanganate 300 OX Ammonium Tetraperoxychromate A - 1 Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special Antimony Compounds Arsenic And Arsenic Compounds Azides Azidocarbonyl Guanidine Barium Ba 2 2 1 W Barium Chlorate Ba(ClO3)2*H2O 13477-00- 1 2 0 OX Barium Oxide (Anhydrous) BaO 1304-28-5 2 3 0 Barium Peroxide BaO2 1304-29-6 0 1 0 OX Benzene C6H6 71-43-2 0 2 3 Benzene Diazonium Chloride Benzotriazole C6H5N3 95-14-7 0 2 1 Benzoyl Peroxide (C6H5CO)2O2 94-36-0 4 1 4 OX Benzyl Alcohol C6H5CH2OH 100-51-6 0 2 1 Bismuth Nitrate Bi(NO3)3*5H2O 10035-06- 3 1 0 OX Borane,Boranes, Diboranes Boron Tribromide 230 W Boron Trifluoride 140 Bromine Pentafluoride Brf5 7789-30-2 3 4 0 W,O Bromine Trifluoride 3 4 0 W,O Butadiene C4H6/CH2=(CH)2=CH 106-99-0 0 2 4 Butenetroil Trinitrate Cadmium and Cadmium Compounds Calcium Nitrate, Anhydrous Ca(NO3)2 -

Website F Repeated Skin Contact Can Cause Thickened Skin And/Or ( Or in Your Facility’S RTK Patchy Areas of Darkening and Loss of Pigment
Right to Know Hazardous Substance Fact Sheet Common Name: ARSENIC TRISULFIDE Synonyms: Arsenic Sesquisulfide; Arsenous Sulfide; King’s Gold CAS Number: 1303-33-9 Chemical Name: Arsenic Sulfide RTK Substance Number: 0162 Date: December 1998 Revision: July 2008 DOT Number: UN 1557 Description and Use EMERGENCY RESPONDERS >>>> SEE BACK PAGE Arsenic Trisulfide is an odorless, yellow or orange, crystalline Hazard Summary powder. It is used in the manufacture of glass, oil cloth, Hazard Rating NJDOH NFPA linoleum, electrical semi-conductors, photoconductors, and HEALTH - 3 fireworks, as a pigment, and in tanning and pesticides. FLAMMABILITY - 0 REACTIVITY - 0 CARCINOGEN POISONOUS GASES ARE PRODUCED IN FIRE DOES NOT BURN Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; Reasons for Citation 4=severe f Arsenic Trisulfide is on the Right to Know Hazardous Substance List because it is cited by OSHA, ACGIH, DOT, f Arsenic Trisulfide can affect you when inhaled and by NIOSH, NTP, DEP, IARC, IRIS, NFPA and EPA. passing through the skin. f This chemical is on the Special Health Hazard Substance f Arsenic Trisulfide is a CARCINOGEN. HANDLE WITH List. EXTREME CAUTION. f Skin contact can cause irritation, burns, rash and loss of pigment f Eye contact can cause irritation and burns. f Inhaling Arsenic Trisulfide can irritate the nose and throat and can cause an ulcer or hole in the “bone” (septum) dividing the inner nose. SEE GLOSSARY ON PAGE 5. f Exposure to Arsenic Trisulfide can cause weakness, poor appetite, headache, and even death. FIRST AID f Arsenic Trisulfide may damage the nervous system and Eye Contact the liver.