Absbookiucr.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Touching New Heights Editor’S Note
ENSEMBLEENSEMBLE Volume 4 (1) | January – February 2016 Newsletter of the Indo-French Centre for the Promotion of Advanced Research India-France Relations Touching New Heights editor’s note Dear Readers, India and France are witnessing the emergence of a new era of collaborative efforts between the two countries in various sectors. In November/December 2015, France hosted a successful global scale diplomatic event for adopting an agreement by many countries on climate change where India was also an active participant. After the visit of Honourable Prime Minister of India Shri Narendra Modi to France in April 2015, the French President H. E. Dr. Mukesh Kumar Mr. François Hollande visited India in January, 2016 as a Chief Guest on the Director, CEFIPRA occasion of 67th Republic Day of India. During his visit, Indian and French scientific as well as technological communities from academia and industry sectors, joined hands through several Agreements/MoUs signed by the two countries. CEFIPRA had the privilege of hosting the officials / signatories of three of such Agreements/MoU signed on 25 January 2016. CEFIPRA, since its evolution as a unique institutional platform and collaborative mechanism, is contributing through its various interventions in a diverse range of S&T domains. These collaborative efforts are making it possible to generate significant knowledge that has a potential to translate discovery science into solution science. I sincerely wish that all these MoUs will create new pathways to further strengthen the Indo-French collaborative research efforts. inside Editor-in-Chief Dr. Mukesh Kumar ii | editor’s note GD Birla Award x | Director, CEFIPRA Dr. -

M Ethods in P Harmacology and T Oxicology
M ETHODS IN P HARMACOLOGY AND T OXICOLOGY Series Editor Y. James Kang Department of Pharmacology and Toxicology, University of Louisville Louisville, KY, USA For further volumes: http://www.springer.com/series/7653 Methods in Pharmacology and Toxicology publishes cutting-edge techniques, including meth- ods, protocols, and other hands-on guidance and context, in all areas of pharmacological and toxicological research. Each book in the series offers time-tested laboratory protocols and expert navigation necessary to aid toxicologists and pharmaceutical scientists in labora- tory testing and beyond. With an emphasis on details and practicality, Methods in Pharma- cology and Toxicology focuses on topics with wide-ranging implications on human health in order to provide investigators with highly useful compendiums of key strategies and approaches to successful research in their respective areas of study and practice. In Silico Modeling of Drugs Against Coronaviruses Computational Tools and Protocols Edited by Kunal Roy Drug Theoretics and Cheminformatics Laboratory, Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India Editor Kunal Roy Drug Theoretics and Cheminformatics Laboratory, Department of Pharmaceutical Technology Jadavpur University Kolkata, India ISSN 1557-2153 ISSN 1940-6053 (electronic) Methods in Pharmacology and Toxicology ISBN 978-1-0716-1365-8 ISBN 978-1-0716-1366-5 (eBook) https://doi.org/10.1007/978-1-0716-1366-5 © The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature 2021 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. -

Folio Id Name 1 Add 1 Add 2 Add 3 City Pin Net Div War No *00005 Balasubramaniam M 6, East 9Th Street, Bigstone Gap Virginia, U S a 24219
UNCLAIMED DIVIDEND FOLIO_ID NAME_1 ADD_1 ADD_2 ADD_3 CITY PIN NET_DIV WAR_NO *00005 BALASUBRAMANIAM M 6, EAST 9TH STREET, BIGSTONE GAP VIRGINIA, U S A 24219. 0 1750.00 013751 *00006 JAGANATHAN R FLAT NO 13 STEVE BIKO ROAD LONDON N7 M J F 0 700.00 014079 *00017 ARCHIBALD FRANCIS RASQUINHA THE CHARTERED BANK P O BOX 29 DOHA QATAR A GULF 0 5474.00 014007 *00031 KRISHNASWAMY RANGANATHAN SUKUMAR MIDEAST CONSTRUCTIONS LTD P O BOX 3325 DOHA QATAR 0 350.00 013942 *00047 BALASUBRAMANIAM G K GRAY MACKENZIE & CO LTD POST BOX NO 186 SHARJAH U A E 0 1400.00 013943 *00066 ZOYEB FIDAHUSAIN RAMPURAWALA POST BOX 745 DUBAI U A E 0 350.00 013944 *00083 BALARAJ SAMSON WALTER GNANAPRAKASA 4-D AYYASAMY STREET PUTHUR TRICHIRAPALLI 620 017 620017 350.00 006020 *00104 MOHAMMAD H MUZAFER NO 73 VELACHEERY ROAD GUINDY CHENNAI 600 032 600032 1050.00 004910 *00105 SAMEER MUZAFER NO 73 VELACHEERY ROAD GUINDY CHENNAI 600 032 600020 1050.00 004599 *00138 SREENIVASAN KRISHNA 409 EASTLAKE AVE E APT 302 SEATTLE WA 98109 U S A 0 350.00 013946 *00140 JULIET ASOKAN P O BOX NO 3997 RUWI POSTAL CODE 112 SULTANATE OF OMAN 0 1400.00 014085 *00141 DEV ASOKAN GENERAL MANAGER AL INARA ENTERPRISES P O BOX NO 3997 RUWI POSTAL CODE 112 SULTANATE OF OMAN 0 2100.00 014086 *00143 SIVAKUMAR S 1398/99-2C VISHAKA 15TH MAIN ROAD ANNA NAGAR CHENNAI 600 040 600040 1708.00 005176 *00146 SAMPATH DEVNATH ENGR KASSEM DARWISH FAKHROO & SONS P O BOX 3898 DOHA QATAR 0 700.00 014087 *00165 MADHAVAN KARUNA KARAN MANUFACTURING MANAGER PUGODA MILLS PUGODA SRI LANKA 0 1750.00 013947 *00169 SALAHUDDIN SYED -

Protein Folding and Dynamics at NCBS, Bengaluru
The Third International Symposium on Protein Folding and Dynamics at NCBS, Bengaluru ORGANIZERS CONFIRMED SPEAKERS Jayant B. Udgaonkar Douglas Barrick Johns Hopkins University, USA NCBS, Bengaluru Paula Booth King’s College London, UK Patricia Clark Notre Dame University, USA C. Robert Matthews Jane Clarke University of Cambridge, UK University of Massachusetts, USA Lila Gierasch University of Massachusetts, USA Shachi Gosavi NCBS, Bengaluru Yuji Goto Osaka University, Japan Gilad Haran Weizmann Institute of Science, Israel Michael Harms University of Oregon, USA Hagen Hofmann Weizmann Institute of Science, Israel Gerhard Hummer Max Planck Institute of Biophysics, Germany th th Thomas Kiefhaber University of Halle, Germany 8 -11 Jooyoung Lee KIAS, South Korea R. Mahalakshmi IISER, Bhopal Samrat Mukhopadhyay IISER, Mohali November, 2016 Sudipta Maiti TIFR, Mumbai Athi Naganathan IIT, Madras NCBS, Bengaluru Rohit Pappu Washington University, USA Sheena Radford University of Leeds, UK Govardhan Reddy IISc , Bengaluru Catherine Royer Rensselaer Polytechnic Institute, USA Registration deadline Tobin Sosnick University of Chicago, USA Hideki Taguchi Tokyo Institute of Technology, Japan July 1, 2016 Tahei Tahara RIKEN, Japan Satoshi Takahashi Tohoku University, Japan Michael Woodside University of Alberta, Canada Tae-Young Yoon KAIST, South Korea The symposium will consist of talks by students and postdoctoral fellows, discussions led by faculty members but driven by students, poster sessions, as well as talks by leaders in the of Protein Folding and Dynamics . For more information and to apply visit National Centre for Biological Sciences https://events.ncbs.res.in/event/third-international- Tata Institute of Fundamental Research symposium-protein-folding-and-dynamics GKVK, Bellary Road, Bengaluru -560065 THIRD INTERNATIONAL PROTEIN FOLDING Sponsors SYMPOSIUM THIRD INTERNATIONAL PROTEIN FOLDING Sponsors SYMPOSIUM Daily THIRD INTERNATIONAL PROTEIN Schedule FOLDING SYMPOSIUM Tuesday, November 8, 2016 08:00 – 08:45 Registration 08:45 – 09:00 Welcome by Jayant B. -
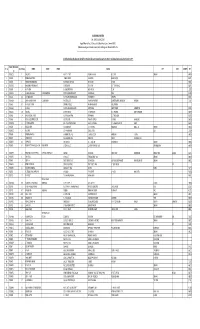
SICAGEN INDIA LIMITED List of Shareholders Who Have Not
SICAGEN INDIA LIMITED CIN: L74900TN2004PLC053467 Regd. Office: 4th Floor, SPIC House, No.88, Mount Road, Guindy, Chennai-600032. Website: www.sicagen.com E-mail: [email protected] Phone: 044 4075 4075. --------------------------------------------------------------------------------------------------------------------------------------- List of Shareholders who have not claimed their Dividends for seven consecutive years since 2010-11 and whose shares are due for Transfer to IEPF FOLIO / DP ID Client Sl. No No. of Shares NAME1 NAME2 NAME3 Address CITY STATE COUNTRY PIN ID 1 '00045015 1 SAROJA P S NO.I 5TH STREET VAISHNAVI NAGAR R.C.C. POST CHENNAI 600109 2 '00045198 14 RAMESH WADHWANI JAGMAL CHOWK MAIN ROAD BILASPUR (C G) 495004 3 '00045378 4 VIHARI BHIKHABHAI DESAI BHAVANIDAS NI KHADKI DESAI VAGO NADIAD 387001 4 '00045451 14 MANUBHAI FAKIRBAHI NAYEE AT SARDARPUR TA VIJAPUR DIST MEHSANA (NG) 382870 5 '00045497 14 ANITA VERMA B-136/4 STREET NO-8 BHAJANPURA DELHI 110053 6 '00045512 14 SUDHIR DUPPALIWAR DIPAK DESHPANDE 44/74 NAVSAHYADRI SOCIETY KARVE NAGAR PUNE 411052 7 '00045528 28 NATHMAL MODI NATHMAL MAHENDRA KUMAR MAIN MARKET CHIRAWA 333026 8 '00045539 25 SHYAM SUNDER TANTIA GEETHA TANTIA FLAT-5C BLOCK-D GANPATHI APARTMENT 21 KHETRA MITRA LANE SALKIA HOWRAH 711106 9 '00045566 50 MUTHUKRISHNAN K SATHANUR VILLEGE VIKKARAWANDI POST VILLUPURAM-5 10 '00045633 4 MURALI G NO 19 PALANI AMMAL ILLAM AYYPPA NAGAR CHETTY STREET COIMBATORE 641001 11 '00045655 100 YASODHA AT POST GUNBALA TK MUTHUKOOR DIST WALYOND ANDHRA PRADESH 508277 12 -

Unpaid Dividend Warrant for the Year 2016
Unpaid Dividend Warrant for the year 2016 FOLIO_ID NAME_1 ADD_1 ADD_2 ADD_3 CITY PIN NET_DIV WAR_NO CHQ_NO DD_DATE 10003 THOMAS GEORGE RAJAN COTTAGE MUNDAKKAL [MIDDLE] QUILON KERALA 691001 600.00 008269 0 10006 THOMAS M V NO 7 NEW KODAMBAKKAM ROAD MADRAS-600 029 600029 6000.00 0 1001 BASANTH KUMAR SUBUDHI AT /PO BINKA BOLANGIR DIST BOLANGIR DIST ORRISA 767019 300.00 008597 0 10012 THRINADHA RAO DUNNA INCOMETAX OFFICE 9-15-10 C B M COMPOUND VISAKHAPATNAM AP 0 300.00 002432 0 10014 THRIPURA BALU N 3/82 AMMAVARISALA STREET RAJAMPET 516 115 CUDDAPAH 516115 600.00 004955 0 10015 THRIPURAMBA T PARTHA APARTMENTS 12/6 VEMBULI AMMAN KOIL STREET VIRUGAMBAKKAM WEST K K NAGAR MADRAS 600 078 600078 600.00 007281 0 10018 THULASIMANI M 56 YANAM VENKATASALA PILLAI ST PONDICHERRY 605 001 605001 600.00 007549 0 10019 THULASIMANI M 56 YANAM VENKATASALA PILLAI ST PONDICHERRY 605001 605001 7200.00 001833 0 1002 BASANTI MISHRA D-117 KOEL NAGAR ROURKELA SUNDARGARH ORISSA 769014 600.00 008613 0 10020 THULASIMANI M 56 YANAM VENKATASALA PILLAI STREET YANAM PONDICHERRY 605001 300.00 007550 0 10023 THUMALAPALLI SESHAGIRI RAO 10-2-289/59 SHANTHI NAGAR COLONY HYDERABAD ANDHRA PRADESH 500028 300.00 004174 0 10025 TIKAM CHAND JAIN SUREKHA CLOTH MERCHANTS POST DORNAKAL DIST WARANGAL ANDHRA PRADESH 506381 300.00 004698 0 10029 TIRUPATHY SWAMY GUDALA 5-16-34 SARWABATLA VARI STREET KAVALI-524 201 524201 600.00 005702 0 10030 TIRUPATI VYANKATRAMANAMDEO JOSHI C/O SHRI N V JOSHI 354 SOMWAR KARAD 415 110 415110 600.00 003666 0 10035 TIWARI B L NO 83-A/63 TIWARI HOUSE JUHI KANPUR 208 014 U P 208014 1200.00 002743 0 10042 TRIBHOOVANPAL GOVERDHANDAS S DUGAR BROTHERS & CO. -

Activity Report 2013–2014
ACTIVITY REPORT 2013–2014 L V Prasad Eye Institute Copyright © 2014 All rights reserved EDITORS: Dr Sreedevi Yadavalli, Aravind Chandarlapati DESIGN: Kartheek Koyinni, Y Yedukondalu ASSISTANCE: V Srinivasa Raju, G Rekha Sruthi, N Jayalaxmi and Lakshmi Sakuntala PHOTOGRAPHY: SBN Chary and Sandeep Roy; LVPEI Archives; Grateful thanks to Vicky Roy DONOR RELATIONS: Sam J Balasundaram PRINTERS: TOTEM Advertising & PR Pvt. Ltd. Department of Communications Level 4 L V Prasad Eye Institute Kallam Anji Reddy Campus L V Prasad Marg, Banjara Hills Hyderabad - 500034, India Ph: +91 40 30612445, +91 40 30612446 Email: [email protected], [email protected] Contents The LVPEI Network 02 The Year at a Glance 04 Our Team 06 Boards of Management 08 Foreword 09 Awards and Honours 10 Breaking New Ground 13 Network News 16 Campus News 22 Kallam Anji Reddy Campus, Hyderabad Bhubaneswar Campus GMR Varalakshmi Campus, Visakhapatnam Kode Venkatadri Chowdary Campus, Vijayawada Patient Care Services 44 Vision Rehabilitation 50 Eye Banking 58 Product Development 62 Gullapalli Pratibha Rao International Centre for Advancement of Rural Eye care 64 Academy of Eye Care Education 78 Prof Brien Holden Eye Research Centre 93 Alumni News 121 Our Support 122 Abbreviations Used KAR - Kallam Anji Reddy Campus RIEB - Ramayamma International Eye Bank GMRV - GMR Varalakshmi Campus GPR ICARE - Gullapalli Pratibha Rao International KVC - Kode Venkatadri Chowdary Campus Centre for Advancement of Rural Eye care THE LVPEI NETWORK Centre of Excellence Centre of Excellence -

International
December 2013 Current Affairs Study Material INTERNATIONAL Asian Countries Top OECD‟s PISA survey of Global Education According to the Paris-based Organisation for Economic Cooperation and Development (OECD) latest survey report , which evaluates the knowledge and skills of the world‘s 15-year-olds in 65 countries and has been released on 3 December. The OECD‘s PISA (Programme for International Student Assessment) 2012 tested more than 510,000 students in 65 countries and economies on maths, reading and science. The main focus was on maths. Highlights of the Survey Report :- * Asian nations cemented their top positions in an eagerly awaited report on global education , as their students continued to outshine western counterparts in maths, science and reading. * Shanghai again ranked first in maths, science and reading . * Singapore, Hong Kong, Taiwan and South Korea rounded out the top five in maths skills. * Already strong performers in 2009, Shanghai, Hong Kong and Singapore continued to improve their performances in the three categories. * The report highlighted Italy, Poland and Portugal for showing improvements in maths skills since the last survey ,but noted drops in Sweden and Finland. * Peru ranked at the bottom of the list in all three categories. * 23 per cent of students in OECD countries, and 32 per cent overall, failed to master the simplest maths problems.Boys performed better than girls in maths. They scored higher in 37 out of the 65 countries and economies, while girls outperformed boys in 5 countries. * Across OECD countries, 8.4% of students are top performers in reading. Shanghai-China has the largest proportion of top performers – 25.1%. -

The Year Book 2019
THE YEAR BOOK 2019 INDIAN ACADEMY OF SCIENCES Bengaluru Postal Address: Indian Academy of Sciences Post Box No. 8005 C.V. Raman Avenue Sadashivanagar Post, Raman Research Institute Campus Bengaluru 560 080 India Telephone : +91-80-2266 1200, +91-80-2266 1203 Fax : +91-80-2361 6094 Email : [email protected], [email protected] Website : www.ias.ac.in © 2019 Indian Academy of Sciences Information in this Year Book is updated up to 22 February 2019. Editorial & Production Team: Nalini, B.R. Thirumalai, N. Vanitha, M. Venugopal, M.S. Published by: Executive Secretary, Indian Academy of Sciences Text formatted by WINTECS Typesetters, Bengaluru (Ph. +91-80-2332 7311) Printed by Lotus Printers Pvt. Ltd., Bengaluru CONTENTS Page Section A: Indian Academy of Sciences Memorandum of Association ................................................... 2 Role of the Academy ............................................................... 4 Statutes .................................................................................. 7 Council for the period 2019–2021 ............................................ 18 Office Bearers ......................................................................... 19 Former Presidents ................................................................... 20 Activities – a profile ................................................................. 21 Academy Document on Scientific Values ................................. 25 The Academy Trust ................................................................. 33 Section B: Professorships -

Annual Report 2014-15
Annual Report 2014-15 Indo-French Centre for the Centre Franco-Indien pour la Promotion of Advanced Research Promotion de la Recherche Avancée Annual Report | 2014-15 1 Indo-French Centre for the Promotion of Advanced Research Annual Report 2014-15 From the Director’s Desk am immensely delighted to present The innovation programme has gained pace the Annual Report of the Indo-French with the initiation of new projects under I Centre for the Promotion of Advanced CEFIPRA’s collaboration with Saint Gobain Research (IFCPAR / CEFIPRA) for the year Research India (SGRI). The discussions 2014-2015. I would like to congratulate for signing an MoU between CEFIPRA my predecessor, Dr. Debapriya Dutta, and Airbus Group have also matured for successfully steering the activities of considerably during this period. Research CEFIPRA as per the mandate given by its projects under the programme on Red Governing Body. The work presented in Biotechnology, involving BIRAC (a Not-for- this report has been mostly initiated and/ or accomplished during his tenure. Biotechnology, Govt. of India) and Bpifrance Profit(a French Company Public set Investment up by Department Bank) are of CEFIPRA has continued its tradition of focusing upto pre-commercialization stage. strengthening the Indo-French Science, Technology and Innovation system Seminars and workshops are an integral by supporting various research and part of CEFIPRA’s bouquet of programmatic development projects. While continuing offerings for the scientists of both the to support research in basic and applied countries. In this period, in addition sciences through its Collaborative to other seminars/workshops focusing various S&T topics, a seminar dedicated to Industry Academia Research Development ScientificProgramme, Research CEFIPRA Programmeis taking steadyand achievements of women PIs in CEFIPRA steps towards creating a solution oriented highlightingsupported projects the scientific was also contribution organized. -

OUTCOME ANALYSIS of RESEARCH PROJECTS COMPLETED DURING 2011 to 2015 UNDER COLLABORATIVE SCIENTIFIC RESEARCH PROGRAMME
OUTCOME ANALYSIS OF RESEARCH PROJECTS COMPLETED DURING 2011 to 2015 UNDER COLLABORATIVE SCIENTIFIC RESEARCH PROGRAMME INDO-FRENCH CENTRE FOR THE CENTRE FRANCO-INDIEN POUR LA PROMOTION OF ADVANCED RESEARCH PROMOTION DE LA RECHERCHE (IFCPAR) AVANCEE (CEFIPRA) FOREWORD FROM THE DIRECTOR It gives me immense pleasure to present this report which contains the outcome analysis of the projects completed during 2011 to 2015 under the Collaborative Scientific Research Programme of CEFIPRA. It is a key programme which has been instrumental in supporting basic and applied research between India and France in advanced areas of Science & Technology. This programme has significant impact in promoting collaborative research in both countries. The joint publications emerged from the CEFIPRA supported projects under this programme covers the ten major thrust areas and contributed to strengthen the research activities between the two countries. The evaluation of this programme indicates that it has played a major role in linking scientists and the research institutions of both countries and contributing towards development of skill in young researchers. The successful completion of ninety four projects during the period of five years, 2011 to 2015 has indicated growth in developing knowledge base of scientific community of both countries and addressing research challenges, areas of global concerns and finding solutions. A short summary of significant achievements is given in Annexure-I. I hope that the information provided in this report will be beneficial for the administrators and policy makers of both countries in shaping future interventional policies in line with Collaborative Scientific Research Programme of the Centre which is contributing in strengthening Indo-French collaboration. -

Annual Report 2008-2009
ANNUAL REPORT 2008-2009 Zebrafish http://genome.igib.res.in wild-type straiGen nome Council of Scientific and Industrial Research New Delhi D R U G D I S C O O V P E E R Y N S O U R C E e e g g Lavendar a a Cultivation p Asiatic in J&K p Lily in r r Himachal e e Pradesh v v OSDD-a CSIR-led o o initiative for affordable C C Herbal healthcare Formulation r r Zebra Fish for prostate o o cancer f f s s Photonic Crystal d d Fibre n n e e Soleckshaw g g e e L L With compliments of Prof. S.K. Brahmachari Director General Council of Scientific & Industrial Research New Delhi e e g g Lavendar a a Cultivation p Asiatic in J&K p Lily in r r Himachal e e Pradesh v v OSDD-a CSIR-led o o initiative for affordable C C Herbal healthcare Formulation r r Zebra Fish for prostate o o cancer f f s s Photonic Crystal d d Fibre n n e e Soleckshaw g g e e L L ANNUAL REPORT 2008-2009 Council of Scientific and Industrial Research Anusandhan Bhawan, 2 Rafi Marg, New Delhi - 110 001 www.csir.res.in l l a a n n o o i i t t a a s s i i n n SOCIETY a a President : Prime Minister g g e Vice President : Minister of Science & Technology e r r r r u u O O t t c c R R I I u u GOVERNING r r S S t t BODY C S C S ADVISORY DIRECTOR PERFORMANCE BOARD GENERAL APPRAISAL BOARDS CSIR HQ MANAGEMENT LABORATORIES RESEARCH COUNCIL (37) COUNCIL Head : Director Contents Page No.