Comparative Network Analysis of Patients with Non-Small Cell Lung Cancer and Smokers for Representing Potential Therapeutic Targ
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multiple Activities of Arl1 Gtpase in the Trans-Golgi Network Chia-Jung Yu1,2 and Fang-Jen S
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 1691-1699 doi:10.1242/jcs.201319 COMMENTARY Multiple activities of Arl1 GTPase in the trans-Golgi network Chia-Jung Yu1,2 and Fang-Jen S. Lee3,4,* ABSTRACT typical features of an Arf-family GTPase, including an amphipathic ADP-ribosylation factors (Arfs) and ADP-ribosylation factor-like N-terminal helix and a consensus site for N-myristoylation (Lu et al., proteins (Arls) are highly conserved small GTPases that function 2001; Price et al., 2005). In yeast, recruitment of Arl1 to the Golgi as main regulators of vesicular trafficking and cytoskeletal complex requires a second Arf-like GTPase, Arl3 (Behnia et al., reorganization. Arl1, the first identified member of the large Arl family, 2004; Setty et al., 2003). Yeast Arl3 lacks a myristoylation site and is an important regulator of Golgi complex structure and function in is, instead, N-terminally acetylated; this modification is required for organisms ranging from yeast to mammals. Together with its effectors, its recruitment to the Golgi complex by Sys1. In mammalian cells, Arl1 has been shown to be involved in several cellular processes, ADP-ribosylation-factor-related protein 1 (Arfrp1), a mammalian including endosomal trans-Golgi network and secretory trafficking, lipid ortholog of yeast Arl3, plays a pivotal role in the recruitment of Arl1 droplet and salivary granule formation, innate immunity and neuronal to the trans-Golgi network (TGN) (Behnia et al., 2004; Panic et al., development, stress tolerance, as well as the response of the unfolded 2003b; Setty et al., 2003; Zahn et al., 2006). -

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Survival-Associated Metabolic Genes in Colon and Rectal Cancers
Survival-associated Metabolic Genes in Colon and Rectal Cancers Yanfen Cui ( [email protected] ) Tianjin Cancer Institute: Tianjin Tumor Hospital https://orcid.org/0000-0001-7760-7503 Baoai Han tianjin tumor hospital He Zhang tianjin tumor hospital Zhiyong Wang tianjin tumor hospital Hui Liu tianjin tumor hospital Fei Zhang tianjin tumor hospital Ruifang Niu tianjin tumor hospital Research Keywords: colon cancer, rectal cancer, prognosis, metabolism Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117478/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/42 Abstract Background Uncontrolled proliferation is the most prominent biological feature of tumors. To rapidly proliferate and maximize the use of available nutrients, tumor cells regulate their metabolic behavior and the expression of metabolism-related genes (MRGs). In this study, we aimed to construct prognosis models for colon and rectal cancers, using MRGs to indicate the prognoses of patients. Methods We rst acquired the gene expression proles of colon and rectal cancers from the TCGA and GEO database, and utilized univariate Cox analysis, lasso regression, and multivariable cox analysis to identify MRGs for risk models. Then GSEA and KEGG functional enrichment analysis were utilized to identify the metabolism pathway of MRGs in the risk models and analyzed these genes comprehensively using GSCALite. Results Eight genes (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR) in the colon cancer risk model and six genes (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) in the rectal cancer risk model were identied successfully. Multivariate Cox analysis indicated that the models predicted overall survival accurately and independently for patients with colon or rectal cancer. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -
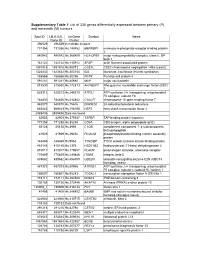
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Supplementary Table S2
1-high in cerebrotropic Gene P-value patients Definition BCHE 2.00E-04 1 Butyrylcholinesterase PLCB2 2.00E-04 -1 Phospholipase C, beta 2 SF3B1 2.00E-04 -1 Splicing factor 3b, subunit 1 BCHE 0.00022 1 Butyrylcholinesterase ZNF721 0.00028 -1 Zinc finger protein 721 GNAI1 0.00044 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 GNAI1 0.00049 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 PDE1B 0.00069 -1 Phosphodiesterase 1B, calmodulin-dependent MCOLN2 0.00085 -1 Mucolipin 2 PGCP 0.00116 1 Plasma glutamate carboxypeptidase TMX4 0.00116 1 Thioredoxin-related transmembrane protein 4 C10orf11 0.00142 1 Chromosome 10 open reading frame 11 TRIM14 0.00156 -1 Tripartite motif-containing 14 APOBEC3D 0.00173 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D ANXA6 0.00185 -1 Annexin A6 NOS3 0.00209 -1 Nitric oxide synthase 3 SELI 0.00209 -1 Selenoprotein I NYNRIN 0.0023 -1 NYN domain and retroviral integrase containing ANKFY1 0.00253 -1 Ankyrin repeat and FYVE domain containing 1 APOBEC3F 0.00278 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F EBI2 0.00278 -1 Epstein-Barr virus induced gene 2 ETHE1 0.00278 1 Ethylmalonic encephalopathy 1 PDE7A 0.00278 -1 Phosphodiesterase 7A HLA-DOA 0.00305 -1 Major histocompatibility complex, class II, DO alpha SOX13 0.00305 1 SRY (sex determining region Y)-box 13 ABHD2 3.34E-03 1 Abhydrolase domain containing 2 MOCS2 0.00334 1 Molybdenum cofactor synthesis 2 TTLL6 0.00365 -1 Tubulin tyrosine ligase-like family, member 6 SHANK3 0.00394 -1 SH3 and multiple ankyrin repeat domains 3 ADCY4 0.004 -1 Adenylate cyclase 4 CD3D 0.004 -1 CD3d molecule, delta (CD3-TCR complex) (CD3D), transcript variant 1, mRNA. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Genome-Wide Meta-Analysisunravels Novel Interactionsbetween
Supplementary Information Genome-wide Meta-analysisUnravels Novel Interactionsbetween Magnesium Homeostasis andMetabolic Phenotypes Table of contents: Supplementary Methods Supplementary Figures 1-10 Supplementary Tables 1-8 Supplementary References 1 SUPPLEMENTARY METHODS GWAS Cohorts CoLaus is a population-based cohort with baseline examination conducted between 2003 and 2006. It includes 6,184 individuals of European descent aged 35-75 years randomly selected from the registry of the city of Lausanne 1. The CROATIA-Vis study, Croatia, is a family- based, cross-sectional study in the isolated island of Vis that included 1,056 examinees aged 18-93. Blood samples were collected in 2003 and 2004 2. The CROATIA-Korcula study, Croatia, is a family-based, cross-sectional study in the isolated island of Korcula that included 965 examinees aged 18-95. Blood samples were collected in 2007 3. The CROATIA-Split study, Croatia, is population-based, cross-sectional study in the Dalmatian city of Split that so far includes 1,012 examinees aged 18-95. Blood samples were collected in 2009 -2011 4. The Lothian Birth Cohort 1936 (LBC1936) consists of 1,091 relatively healthy older participants, most of whom took part in the Scottish Mental Survey of 1947 at the age of about 11 years. At a mean age of 69.5 years (SD 0.8) they were recruited to a study investigating influences on cognitive ageing 5. A second wave of cognitive and physical testing occurred at approximately 73 years of age at which time a urine sample was collected 5, 6. The INGI-Val Borbera population is a collection of 1,785 genotyped samples (18-102 years) collected in the Val Borbera Valley, a geographically isolated valley located within the Appennine Mountains in Northwest Italy 7. -

Novel and Highly Recurrent Chromosomal Alterations in Se´Zary Syndrome
Research Article Novel and Highly Recurrent Chromosomal Alterations in Se´zary Syndrome Maarten H. Vermeer,1 Remco van Doorn,1 Remco Dijkman,1 Xin Mao,3 Sean Whittaker,3 Pieter C. van Voorst Vader,4 Marie-Jeanne P. Gerritsen,5 Marie-Louise Geerts,6 Sylke Gellrich,7 Ola So¨derberg,8 Karl-Johan Leuchowius,8 Ulf Landegren,8 Jacoba J. Out-Luiting,1 Jeroen Knijnenburg,2 Marije IJszenga,2 Karoly Szuhai,2 Rein Willemze,1 and Cornelis P. Tensen1 Departments of 1Dermatology and 2Molecular Cell Biology, Leiden University Medical Center, Leiden, the Netherlands; 3Department of Dermatology, St Thomas’ Hospital, King’s College, London, United Kingdom; 4Department of Dermatology, University Medical Center Groningen, Groningen, the Netherlands; 5Department of Dermatology, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands; 6Department of Dermatology, Gent University Hospital, Gent, Belgium; 7Department of Dermatology, Charite, Berlin, Germany; and 8Department of Genetics and Pathology, Rudbeck Laboratory, University of Uppsala, Uppsala, Sweden Abstract Introduction This study was designed to identify highly recurrent genetic Se´zary syndrome (Sz) is an aggressive type of cutaneous T-cell alterations typical of Se´zary syndrome (Sz), an aggressive lymphoma/leukemia of skin-homing, CD4+ memory T cells and is cutaneous T-cell lymphoma/leukemia, possibly revealing characterized by erythroderma, generalized lymphadenopathy, and pathogenetic mechanisms and novel therapeutic targets. the presence of neoplastic T cells (Se´zary cells) in the skin, lymph High-resolution array-based comparative genomic hybridiza- nodes, and peripheral blood (1). Sz has a poor prognosis, with a tion was done on malignant T cells from 20 patients. disease-specific 5-year survival of f24% (1). -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin