Competitive Displacement Between Bemisia Tabaci MEAM1 and MED and Evidence for Multiple Invasions of MED
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CIFI Holdings (Group) Co. Ltd. 旭 輝 控 股(集 團)有 限
Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this announcement, make no representation as to its accuracy or completeness, and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this announcement. CIFI Holdings (Group) Co. Ltd. 旭輝控股(集團)有限公司 (Incorporated in the Cayman Islands with limited liability) (Stock Code: 00884) ANNOUNCEMENT OF UNAUDITED INTERIM RESULTS FOR THE SIX MONTHS ENDED 30 JUNE 2020 2020 INTERIM RESULTS HIGHLIGHTS • Recognized revenue increased by 11.3% year-on-year to RMB23.02 billion • Core net profit increased by 11.2% year-on-year to RMB3,194 million, with core net profit margin at 13.9%. Stable gross profit of approximately RMB5,901 million • Declared interim dividend of RMB9.8 cents (or equivalent to HK11 cents) per share, increased by 10% year-on-year • Contracted sales amounted to RMB80.7 billion with cash collection ratio from property sales achieved over 95% • As at 30 June 2020, net debt-to-equity ratio decreased by 2.4 percentage points to 63.2% comparing with that as at 31 December 2019. Abundant cash on hand of RMB59.4 billion • As at 30 June 2020, weighted average cost of indebtedness decreased by 0.4 percentage point to 5.6% comparing with that as at 31 December 2019 – 1 – INTERIM RESULTS The board of directors (the “Board”) of CIFI Holdings (Group) Co. Ltd. (the “Company”) is pleased to announce the unaudited consolidated results of the -
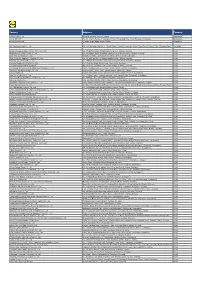
Factory Address Country
Factory Address Country Durable Plastic Ltd. Mulgaon, Kaligonj, Gazipur, Dhaka Bangladesh Lhotse (BD) Ltd. Plot No. 60&61, Sector -3, Karnaphuli Export Processing Zone, North Potenga, Chittagong Bangladesh Bengal Plastics Ltd. Yearpur, Zirabo Bazar, Savar, Dhaka Bangladesh ASF Sporting Goods Co., Ltd. Km 38.5, National Road No. 3, Thlork Village, Chonrok Commune, Korng Pisey District, Konrrg Pisey, Kampong Speu Cambodia Ningbo Zhongyuan Alljoy Fishing Tackle Co., Ltd. No. 416 Binhai Road, Hangzhou Bay New Zone, Ningbo, Zhejiang China Ningbo Energy Power Tools Co., Ltd. No. 50 Dongbei Road, Dongqiao Industrial Zone, Haishu District, Ningbo, Zhejiang China Junhe Pumps Holding Co., Ltd. Wanzhong Villiage, Jishigang Town, Haishu District, Ningbo, Zhejiang China Skybest Electric Appliance (Suzhou) Co., Ltd. No. 18 Hua Hong Street, Suzhou Industrial Park, Suzhou, Jiangsu China Zhejiang Safun Industrial Co., Ltd. No. 7 Mingyuannan Road, Economic Development Zone, Yongkang, Zhejiang China Zhejiang Dingxin Arts&Crafts Co., Ltd. No. 21 Linxian Road, Baishuiyang Town, Linhai, Zhejiang China Zhejiang Natural Outdoor Goods Inc. Xiacao Village, Pingqiao Town, Tiantai County, Taizhou, Zhejiang China Guangdong Xinbao Electrical Appliances Holdings Co., Ltd. South Zhenghe Road, Leliu Town, Shunde District, Foshan, Guangdong China Yangzhou Juli Sports Articles Co., Ltd. Fudong Village, Xiaoji Town, Jiangdu District, Yangzhou, Jiangsu China Eyarn Lighting Ltd. Yaying Gang, Shixi Village, Shishan Town, Nanhai District, Foshan, Guangdong China Lipan Gift & Lighting Co., Ltd. No. 2 Guliao Road 3, Science Industrial Zone, Tangxia Town, Dongguan, Guangdong China Zhan Jiang Kang Nian Rubber Product Co., Ltd. No. 85 Middle Shen Chuan Road, Zhanjiang, Guangdong China Ansen Electronics Co. Ning Tau Administrative District, Qiao Tau Zhen, Dongguan, Guangdong China Changshu Tongrun Auto Accessory Co., Ltd. -

Transmissibility of Hand, Foot, and Mouth Disease in 97 Counties of Jiangsu Province, China, 2015- 2020
Transmissibility of Hand, Foot, and Mouth Disease in 97 Counties of Jiangsu Province, China, 2015- 2020 Wei Zhang Xiamen University Jia Rui Xiamen University Xiaoqing Cheng Jiangsu Provincial Center for Disease Control and Prevention Bin Deng Xiamen University Hesong Zhang Xiamen University Lijing Huang Xiamen University Lexin Zhang Xiamen University Simiao Zuo Xiamen University Junru Li Xiamen University XingCheng Huang Xiamen University Yanhua Su Xiamen University Benhua Zhao Xiamen University Yan Niu Chinese Center for Disease Control and Prevention, Beijing City, People’s Republic of China Hongwei Li Xiamen University Jian-li Hu Jiangsu Provincial Center for Disease Control and Prevention Tianmu Chen ( [email protected] ) Page 1/30 Xiamen University Research Article Keywords: Hand foot mouth disease, Jiangsu Province, model, transmissibility, effective reproduction number Posted Date: July 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-752604/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/30 Abstract Background: Hand, foot, and mouth disease (HFMD) has been a serious disease burden in the Asia Pacic region represented by China, and the transmission characteristics of HFMD in regions haven’t been clear. This study calculated the transmissibility of HFMD at county levels in Jiangsu Province, China, analyzed the differences of transmissibility and explored the reasons. Methods: We built susceptible-exposed-infectious-asymptomatic-removed (SEIAR) model for seasonal characteristics of HFMD, estimated effective reproduction number (Reff) by tting the incidence of HFMD in 97 counties of Jiangsu Province from 2015 to 2020, compared incidence rate and transmissibility in different counties by non -parametric test, rapid cluster analysis and rank-sum ratio. -

CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A Joint Stock Company Incorporated in the People’S Republic of China with Limited Liability) (Stock Code: 2202)
Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this announcement, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this announcement. CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A joint stock company incorporated in the People’s Republic of China with limited liability) (Stock Code: 2202) 2019 ANNUAL RESULTS ANNOUNCEMENT The board of directors (the “Board”) of China Vanke Co., Ltd.* (the “Company”) is pleased to announce the audited results of the Company and its subsidiaries for the year ended 31 December 2019. This announcement, containing the full text of the 2019 Annual Report of the Company, complies with the relevant requirements of the Rules Governing the Listing of Securities on The Stock Exchange of Hong Kong Limited in relation to information to accompany preliminary announcement of annual results. Printed version of the Company’s 2019 Annual Report will be delivered to the H-Share Holders of the Company and available for viewing on the websites of The Stock Exchange of Hong Kong Limited (www.hkexnews.hk) and of the Company (www.vanke.com) in April 2020. Both the Chinese and English versions of this results announcement are available on the websites of the Company (www.vanke.com) and The Stock Exchange of Hong Kong Limited (www.hkexnews.hk). In the event of any discrepancies in interpretations between the English version and Chinese version, the Chinese version shall prevail, except for the financial report prepared in accordance with International Financial Reporting Standards, of which the English version shall prevail. -

Levi Strauss & Co. Factory List
LEVI STRAUSS & CO. FACTORY LIST Published : September 2014 Country Factory name Alternative factory name Address City State/Province Argentina Accecuer SA Juan Zanella 4656 Caseros Buenos Aires Avanti S.A. Coronel Suarez 1544 Olavarría Buenos Aires Best Sox S.A. Charlone 1446 Capital Federal Buenos Aires Buffalo S.R.L. Valentín Vergara 4633 Florida Oeste Buenos Aires Carlos Kot San Carlos 1047 Wilde Buenos Aires CBTex S.R.L. - Cut Avenida de los Constituyentes, 5938 Capital Federal Buenos Aires CBTex S.R.L. - Sew San Vladimiro, 5643 Lanús Oeste Buenos Aires Cooperativa de Trabajo Textiles Pigue Ltda. Altromercato Ruta Nac. 33 Km 132 Pigue Buenos Aires Divalori S.R.L Miralla 2536 Buenos Aires Buenos Aires Estex Argentina S.R.L. Superi, 3530 Caba Buenos Aires Gitti SRL Italia 4043 Mar del Plata Buenos Aires Huari Confecciones (formerly Victor Flores Adrian) Victor Flores Adrian Charcas, 458 Ramos Mejía Buenos Aires Khamsin S.A. Florentino Ameghino, 1280 Vicente Lopez Buenos Aires Ciudad Autonoma de Buenos La Martingala Av. F.F. de la Cruz 2950 Buenos Aires Aires Lenita S.A. Alfredo Bufano, 2451 Capital Federal Buenos Aires Manufactura Arrecifes S.A. Ruta Nacional 8, Kilometro 178 Arrecifes Buenos Aires Materia Prima S.A. - Planta 2 Acasusso, 5170 Munro Buenos Aires Procesadora Centro S.R.L. Francisco Carelli 2554 Venado Tuerto Santa Fe Procesadora Serviconf SRL Gobernardor Ramon Castro 4765 Vicente Lopez Buenos Aires Procesadora Virasoro S.A. Boulevard Drago 798 Pergamino Buenos Aires Procesos y Diseños Virasoro S.A. Avenida Ovidio Lagos, 4650 Rosario Santa Fé Provira S.A. Avenida Bucar, 1018 Pergamino Buenos Aires Spring S.R.L. -

Levi Strauss & Co. Factory List
Levi Strauss & Co. Factory List Published : November 2019 Total Number of LS&Co. Parent Company Name Employees Country Factory name Alternative Name Address City State Product Type (TOE) Initiatives (Licensee factories are (Workers, Staff, (WWB) blank) Contract Staff) Argentina Accecuer SA Juan Zanella 4656 Caseros Accessories <1000 Capital Argentina Best Sox S.A. Charlone 1446 Federal Apparel <1000 Argentina Estex Argentina S.R.L. Superi, 3530 Caba Apparel <1000 Argentina Gitti SRL Italia 4043 Mar del Plata Apparel <1000 Argentina Manufactura Arrecifes S.A. Ruta Nacional 8, Kilometro 178 Arrecifes Apparel <1000 Argentina Procesadora Serviconf SRL Gobernardor Ramon Castro 4765 Vicente Lopez Apparel <1000 Capital Argentina Spring S.R.L. Darwin, 173 Federal Apparel <1000 Asamblea (101) #536, Villa Lynch Argentina TEXINTER S.A. Texinter S.A. B1672AIB, Buenos Aires Buenos Aires <1000 Argentina Underwear M&S, S.R.L Levalle 449 Avellaneda Apparel <1000 Argentina Vira Offis S.A. Virasoro, 3570 Rosario Apparel <1000 Plot # 246-249, Shiddirgonj, Bangladesh Ananta Apparels Ltd. Nazmul Hoque Narayangonj-1431 Narayangonj Apparel 1000-5000 WWB Ananta KASHPARA, NOYABARI, Bangladesh Ananta Denim Technology Ltd. Mr. Zakaria Habib Tanzil KANCHPUR Narayanganj Apparel 1000-5000 WWB Ananta Ayesha Clothing Company Ltd (Ayesha Bangobandhu Road, Tongabari, Clothing Company Ltd,Hamza Trims Ltd, Gazirchat Alia Madrasha, Ashulia, Bangladesh Hamza Clothing Ltd) Ayesha Clothing Company Ltd( Dhaka Dhaka Apparel 1000-5000 Jamgora, Post Office : Gazirchat Ayesha Clothing Company Ltd (Ayesha Ayesha Clothing Company Ltd(Unit-1)d Alia Madrasha, P.S : Savar, Bangladesh Washing Ltd.) (Ayesha Washing Ltd) Dhaka Dhaka Apparel 1000-5000 Khejur Bagan, Bara Ashulia, Bangladesh Cosmopolitan Industries PVT Ltd CIPL Savar Dhaka Apparel 1000-5000 WWB Epic Designers Ltd 1612, South Salna, Salna Bazar, Bangladesh Cutting Edge Industries Ltd. -

Wuxi Apptec Co., Ltd.* 無錫藥明康德新藥開發股份有限公司
THIS CIRCULAR IS IMPORTANT AND REQUIRES YOUR IMMEDIATE ATTENTION If you are in any doubt as to any aspect of this circular or as to the action to be taken, you should consult a stockbroker or other registered dealer in securities, a bank manager, solicitor, professional accountant or other professional adviser. If you have sold or transferred all your shares in WuXi AppTec Co., Ltd.* (無錫藥明康德新藥開發股份有限公司), you should at once hand this circular, together with the enclosed form of proxy, to the purchaser or transferee or to the bank, stockbroker or other agent through whom the sale or transfer was effected for transmission to the purchaser or transferee. Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this circular, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this circular. WUXI APPTEC CO., LTD.* 無錫藥明康德新藥開發股份有限公司 (A joint stock company incorporated in the People’s Republic of China with limited liability) (Stock Code: 2359) (1) WORK REPORT OF THE BOARD OF DIRECTORS FOR THE YEAR 2019; (2) WORK REPORT OF THE SUPERVISORY COMMITTEE FOR THE YEAR 2019; (3) ANNUAL REPORT FOR THE YEAR 2019; (4) FINANCIAL REPORT FOR THE YEAR 2019; (5) PROPOSED 2019 PROFIT DISTRIBUTION PLAN; (6) PROPOSED RE-ELECTION OF DIRECTORS; (7) PROPOSED ELECTION OF EXECUTIVE DIRECTOR; (8) PROPOSED RE-ELECTION OF SHAREHOLDER REPRESENTATIVE SUPERVISORS; -

Feasibility Study on Launching of Shared New Energy Vehicles in Wuxi City
2018 9th International Symposium on Advanced Education and Management (ISAEM 2018) Feasibility Study on Launching of Shared New Energy Vehicles in Wuxi City Feng Xia School of Automotive Technology, Wuxi Vocational Institute of Commerce, Wuxi, Jiangsu, 214153, China [email protected] Keywords: Shared vehicles; Status quo; Development studies Abstract: Shared vehicles not only bring convenience to urban people to use cars, but also meet the needs of "car-free people", and have been recognized among many citizens. This paper discusses the status quo and development of shared vehicles in Wuxi city, and provides reference for the future entrepreneurship of shared vehicles. Through scientific design of research content and reasonable development of practice plan, different objects were investigated and surveyed in the form of field visit and consultation of users, by distribution of survey questionnaire. 1. Introduction High oil prices have led to a shift in many people’s attitudes towards car-sharing, and they tend to join the “car-sharing” team. The number of car-sharing members in the United States, Canada and other countries has increased rapidly, with nearly 100,000 members in the United States. “Car-sharing” is also booming in countries such as the Netherlands, Belgium, Denmark, Italy and the United Kingdom. Major cities in Belgium have set up special parking spaces for shared vehicles like taxis [1]. The government wants the number of “car-sharing members” to triple by 2010. The Dutch capital, Amsterdam, has 310 shared car stops. Germany is considering amending its laws, allowing special parking spaces for shared cars in residential areas. In the future, "car-sharing" could develop into a daily car service that is more flexible in terms of time, more spatial and less expensive than traditional car rental services, according to George Welk, a sociology and automotive expert at the Institute of Climate, Environment and Energy in Upetal, Germany [2]. -

Federal Register/Vol. 84, No. 70/Thursday, April 11, 2019/Rules
14608 Federal Register / Vol. 84, No. 70 / Thursday, April 11, 2019 / Rules and Regulations SUMMARY: This action withdraws the NATIONAL AERONAUTICS AND Background FAA’s interpretation of the special rule SPACE ADMINISTRATION The Unverified List, found in for model aircraft. That interpretation Supplement No. 6 to Part 744 to the 14 CFR Parts 1264 and 1271 no longer is valid or necessary because EAR, contains the names and addresses Congress repealed the special rule for RIN 2700–AE48 of foreign persons who are or have been model aircraft. parties to a transaction, as that such [Document Number NASA–19–003: Docket parties are described in § 748.5 of the DATES: The interpretation published on Number–NASA–2019–0002] June 25, 2014 (79 FR 36172) is EAR, involving the export, reexport, or transfer (in-country) of items subject to withdrawn as of April 11, 2019. Implementation of the Federal Civil Penalties Inflation Adjustment Act and the EAR, and whose bona fides (i.e., FOR FURTHER INFORMATION CONTACT: Adjustment of Amounts for 2019 legitimacy and reliability relating to the Jonathan W. Cross, Regulations end use and end user of items subject Division, Office of the Chief Counsel, Correction to the EAR) BIS has been unable to Federal Aviation Administration, 800 In rule document 2019–06555 verify through an end-use check. BIS Independence Avenue SW, Washington, appearing on pages 13114–13115 in the may add persons to the UVL when BIS DC 20591; telephone 202–267–7173; issue of April 4, 2019, make the or federal officials acting on BIS’s behalf email: [email protected]. -

Suppliers in 2020 Justin.Xlsx
Supplier list Aug-20 On average, Reima has worked with these suppliers for 6,5 years. The list excludes suppliers with annual purchases below twenty thousand Euros. Supplier name Country Address (Production site) 4A Yarn dyeing Ltd Bangladesh Kaicha Bari, Baipail, DEPZ, Savar, Dhaka-1340, Bangladesh Dongyi Shoes & Plastics Co. China Gaoxia Industrial Area, Qingyang District, Jinjiang City, China Anhui Shilin Garment Manufacturing Co. Ltd China Huoshan Economic Development Zone, industrial Area, Huoshan, Liuan, Anhui, China Century Apparels Pvt. Ltd India S.F.No.: 118/2,3/119, Kaniyampoondi, Avinashi-641 663 Tamil Nadu, India Changshu Top-Long Imp.& Exp. Co.Ltd China Cangjing Rd, Yangyuan, Xinzhuang Town, Changshu City Suzhou, Jiangsu Province Dongguan Bestron Industrial Ltd Company China No.5, Hankou 3rd Street, Tudikeng, Shabu Village, Dalang Town Dongguan, Guangdong Dongguan JieYang Footwear Co. Ltd China No.1, Tongfu Road, Shilong Road, SangYuan Industries area, Dongcheng Street, Dongguan, Guangdong, China Fujian Safetech Protection Co., Ltd China No.6 Road East, Tieling Industrial Zone, Zone 1, Block 9, Jingxi Town, Minhou County, Fuzhou, Fujian Province, China Fultex Apparel (Xiamen) Co., Ltd China No.33, Huli Park, Tong An Industrial Cluster, Xiamen 361100, Fujian, China Gartex Exports (India) Private Limited India 50, Lakshmi Buildings, Kangeyam Road, 641 604, Tirupur 641 604, Tamil Nadu, India Great Yo Co., Ltd Taiwan No. 612, Yanxin Street, Yongkang District, Tainan City, Taiwan Haining Yeshi Knitting Co., Ltd. China No. 139, Biyun Road Haining Zhejiang, China Haksan Vi Na Company Limited Vietnam Quarter 3, Tan Dinh Ward, Ben Cat District, Binh Duong, Vietnam Huian Kimsen Footwear Co. -

Voluntary Petition for Garrett Motion Italia S.R.L
20-12230 Doc 1 Filed 09/20/20 Entered 09/20/20 20:19:39 Main Document Pg 1 of 31 )LOOLQWKLVLQIRUPDWLRQWRLGHQWLI\WKHFDVH 8QLWHG6WDWHV%DQNUXSWF\&RXUWIRUWKH BBBBBBBBBBBBBBBBBBBB6RXWKHUQ'LVWULFWRIBBBBBBBBBBBBBBBBB 1HZ <RUN 6WDWH &DVHQXPEHU If known BBBBBBBBBBBBBBBBBBBBBBBBB&KDSWHUBBBBB &KHFNLIWKLVLVDQ DPHQGHGILOLQJ 2IILFLDO)RUP Voluntary Petition for Non-Individuals Filing for Bankruptcy ,IPRUHVSDFHLVQHHGHGDWWDFKDVHSDUDWHVKHHWWRWKLVIRUP2QWKHWRSRIDQ\DGGLWLRQDOSDJHVZULWHWKHGHEWRU¶VQDPHDQGWKHFDVH QXPEHU LINQRZQ )RUPRUHLQIRUPDWLRQDVHSDUDWHGRFXPHQWInstructions for Bankruptcy Forms for Non-Individuals,LVDYDLODEOH 'HEWRU¶VQDPH BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB*DUUHWW 0RWLRQ ,WDOLD 65/ BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB +RQH\ZHOO *DUUHWW ,WDOLD 65/ $OORWKHUQDPHVGHEWRUXVHG BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB LQWKHODVW\HDUV BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB ,QFOXGHDQ\DVVXPHGQDPHV WUDGHQDPHVDQG doing business BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB as QDPHV BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 'HEWRU¶VIHGHUDO(PSOR\HU BBBBBBB±BBBBBBBBBBBBBBBBBBBBBBBBBBB ,GHQWLILFDWLRQ1XPEHU (,1 'HEWRU¶VDGGUHVV 3ULQFLSDOSODFHRIEXVLQHVV 0DLOLQJDGGUHVVLIGLIIHUHQWIURPSULQFLSDOSODFH -

March 31Th, 2020 Today, the Mexican Ministry of Economy Published In
March 31th, 2020 ANTIDUMPING INVESTIGATION ON POLYESTER TEXTURED YARN FROM CHINA AND INDIA Today, the Mexican Ministry of Economy published in the Official Gazette the notice of initiation of the antidumping investigation on Polyester Textured Yarn from China and India. Below, please find relevant information on the antidumping investigation: 1. Petitioner: Akra Polyester, S.A. de C.V. 2. General Product Description: Continuous yarn of textured textile filament or polyester textured yarn (PTY), and correspond to a synthetic continuous multifilament yarn made from polyethylene terephthalate (PET) polyester, either virgin or recycled. 3. Tariff Item (MHTS): 5402.33.01 4. Period of Dumping Investigation: July 1st, 2018 to June 30th, 2019. 5. Period of Injury Analysis: July 1st, 2016 to June 30th, 2019. 6. Normal Value: The Ministry selected the United States as a substitute market economy country for China for the purposes of calculating normal value. As regards India, the Ministry considered the sales prices for consumption in the Indian domestic market. 7. Date to Submit the Questionnaire: May 12th, 2020. 8. Named Exporters: CHINA Air Tiger Express (Asia) Inc. Damco China Ltd. (Shanghai Branch) Jiaye International Town No. 158, 4th Tian An Centre No. 338 Building, Room 2103 Nanjing Road (W) 3-4/F Lushan Road, Jianye DistriCt Zipe Code 200003, Shanghai, China Zip Code 210019, Nanjing, China Enturbo & Jingu Group Inc. Chori (China) Co. Ltd. Wuyuan Bay, Xiamen Yan´an Road (West) Suite 1208 Unit L,8/F., Building 1, Business Internatíonal Trade Center 2201 Operation Center Zip Code 200336, Shanghai, China Zip Code, 361006, Fujian, China Fujian Billion Polymerization Fiber Hangzhou Xianglu Chemical Fiber Co.