Experimental Investigation of the Photophysics of Oregon Green 488 and Its Suitability to Biological Research Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute Fluorescence Quantum Yields Using the FL 6500 Fluorescence Spectrometer
APPLICATION NOTE Fluorescence Spectroscopy Authors: Kathryn Lawson-Wood Steve Upstone Kieran Evans PerkinElmer, Inc. Seer Green, UK Absolute Fluorescence Quantum Yields Using the FL 6500 Introduction Fluorescence quantum yield Fluorescence Spectrometer (ΦF) is a characteristic property of a fluorescent species and is denoted as the ratio of the number of photons emitted through fluorescence, to the number of photons absorbed by the fluorophore (Equation 1). ΦF ultimately relates to the efficiency of the pathways leading to emission of fluorescence (Figure 1), providing the probability of the excited state being deactivated by fluorescence rather than by other competing relaxation processes. The magnitude of ΦF is directly related to the intensity of the observed fluorescence.1 Number of photons emitted through fluorescence ΦF = (1) Number of photons absorbed Fluorescence quantum yield can be measured using two methods: the relative method and the absolute method. In comparison with relative quantum yield measurements, the absolute method is applicable to both solid and solution phase measurements. The ΦF can be determined in a single measurement without the need for a reference standard. It is especially useful for samples which absorb and emit in wavelength regions for which there are no reliable quantum yield standards available.2 The importance of ΦF has been shown in several industries, standard. It is especially useful for samples which absorb and including the research, development and evaluation of audio emit in wavelength regions for which there are no reliable visual equipment, electroluminescent materials (OLED/LED) and quantum yield standards available. This method requires an fluorescent probes for biological assays.3 This application integrating sphere which allows the instrument to collect all of demonstrates the use of the PerkinElmer FL 6500 Fluorescence the photons emitted from the sample. -

Studies on Fluorescence Efficiency and Photodegradation Of
Laser Chemistry, 2002, Vol. 20(2–4), pp. 99–110 Studies on Fluorescence Efficiency and Photodegradation of Rhodamine 6G Doped PMMA Using a Dual Beam Thermal Lens Technique ACHAMMA KURIAN*, NIBU A GEORGE, BINOY PAUL, V. P. N. NAMPOORI and C. P. G. VALLABHAN International School of Photonics, Cochin University of Science & Technology, Cochin 682022, India (Received11September 2002) In this paper we report the use of the dual beam thermal lens technique as a quantitative method to determine absolute fluorescence quantum efficiency and concentration quenching of fluorescence emission from rhodamine 6G doped Poly(methyl methacrylate) (PMMA), prepared with different concentrations of the dye. A comparison of the present data with that reported in the literature indicates that the observed variation of fluorescence quantum yield with respect to the dye concentration follows a similar profile as in the earlier reported observations on rhodamine 6G in solution. The photodegradation of the dye molecules under cw laser excitation is also studied using the present method. Key words: Thermal lens; Fluorescence quantum yield; Photostability; Dye doped polymer 1 INTRODUCTION From the mid 60s, dye lasers have been attractive sources of coherent tunable radiation because of their unique operational flexibility. The salient features of dye lasers are their tunability, with emission from near ultraviolet to the near infrared. High gain, broad spectral bandwidth enabled pulsed and continuous wave operation. Although dyes have been demonstrated to lase in the solid, liquid or gas phases, liquid solutions of dyes in suitable organic * Corresponding author. Catholicate College, Pathanamthitta 689645, India. E-mail: [email protected] 100 A. KURIAN et al. -

Measurement of Fluorescence Quantum Yields on ISS Instrumentation Using Vinci Yevgen Povrozin and Ewald Terpetschnig
TECHNICAL NOTE Measurement of Fluorescence Quantum Yields on ISS Instrumentation Using Vinci Yevgen Povrozin and Ewald Terpetschnig Introduction The quantum yield Q can also be described by the relative rates of the radiative and non-radiative pathways, which deactivate the excited state: kr Q [1 ] kkr nr where and correspond to radiative and non-radiative processes, respectively. In this equation, kr knr knr describes the sum of the rate constants for the various processes that compete with the emission process. These processes include photochemical, and dissociative processes including other, less well characterized changes that result in a return to the ground state with simultaneous dissipation of the energy of the excited state into heat. These latter processes are collectively called non-radiative transitions and two types have been clearly recognized: intersystem crossing and internal conversion. Intersystem crossing related to the radiationless spin inversion of a singlet state (S1) in the excited state into a triplet state (T1). The luminescence quantum yield is also given as the ratio of the number of photons emitted to the number of photons absorbed by the sample: photonsem Q [ 2] photonsabs While measurements of the “absolute” quantum yield do require more sophisticated instrumentation [1], it is easier to determine the “relative” quantum yield of a fluorophore by comparing it to a standard with a known quantum yield. Some of the most common standards are listed in table below: Conditions for Quantum Yield [Q.Y.] Q.Y. Excitation Q.Y. Ref. Standards [nm] [%] Measurements Cy3 4 PBS 540 2 Cy5 27 PBS 620 2 Cresyl Violet 53 Methanol 580 3 1 ISS TECHNICAL NOTE Conditions for Quantum Yield [Q.Y.] Q.Y. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

Two-Photon Excitation Fluorescence Microscopy
P1: FhN/ftt P2: FhN July 10, 2000 11:18 Annual Reviews AR106-15 Annu. Rev. Biomed. Eng. 2000. 02:399–429 Copyright c 2000 by Annual Reviews. All rights reserved TWO-PHOTON EXCITATION FLUORESCENCE MICROSCOPY PeterT.C.So1,ChenY.Dong1, Barry R. Masters2, and Keith M. Berland3 1Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139; e-mail: [email protected] 2Department of Ophthalmology, University of Bern, Bern, Switzerland 3Department of Physics, Emory University, Atlanta, Georgia 30322 Key Words multiphoton, fluorescence spectroscopy, single molecule, functional imaging, tissue imaging ■ Abstract Two-photon fluorescence microscopy is one of the most important re- cent inventions in biological imaging. This technology enables noninvasive study of biological specimens in three dimensions with submicrometer resolution. Two-photon excitation of fluorophores results from the simultaneous absorption of two photons. This excitation process has a number of unique advantages, such as reduced specimen photodamage and enhanced penetration depth. It also produces higher-contrast im- ages and is a novel method to trigger localized photochemical reactions. Two-photon microscopy continues to find an increasing number of applications in biology and medicine. CONTENTS INTRODUCTION ................................................ 400 HISTORICAL REVIEW OF TWO-PHOTON MICROSCOPY TECHNOLOGY ...401 BASIC PRINCIPLES OF TWO-PHOTON MICROSCOPY ..................402 Physical Basis for Two-Photon Excitation ............................ -

Photochemistry
Photochemistry 1.1 Introduction:- Photochemistry is concerned with reactions which are initiated by electronically excited molecules. Such molecules are produced by the absorption of suitable radiation in the visible and near ultraviolet region of the spectrum. Photochemistry is basic to the world we live in with sun as the central figure, the origin of life must have been a photochemical act. Simple gaseous molecules like methane, ammonia and carbon dioxide must have reacted photochemically to synthesize complex organic molecules like proteins and nucleic acids. Photobiology, the photochemistry of biological reactions, is a rapidly developing subject and helps in understanding the phenomenon of photosynthesis, phototaxin, photoperiodism, vision and mutagenic effects of light. The relevance of photochemistry also lies in its varied applications in science and technology. Synthetic organic photochemistry has provided methods for the manufacture of many chemicals which could not be produced by dark reactions. Some industrially viable photochemical syntheses include synthesis of vitamin D2 from ergosterol isolated from yeast, synthesis of caprolactum which is the monomer for Nylon 6, manufacture of cleaning solvents and synthesis of some antioxidants. Photoinitiated polymerization and photopolymerisation are used in photography, lithoprinting and manufacture of printed circuits for the electronic industry. The photophysical phenomena of flourescence and phosphorescence have found varied applications in fluorescent tube lights, TV screens, as luminescent dials for watches, as “optical brighteners” in white dress materials, as paints in advertisement hoardings and so on. Another revolutionary application of electronically excited molecular systems is laser technology. The two main processes, therefore, studied under photochemistry are: 1. Photophysical process 2. Photochemical process 1. -

Lifetime and Fluorescence Quantum Yield of Two Fluorescein-Amino Acid-Based Compounds in Different Organic Solvents and Gold Colloidal Suspensions
chemosensors Article Lifetime and Fluorescence Quantum Yield of Two Fluorescein-Amino Acid-Based Compounds in Different Organic Solvents and Gold Colloidal Suspensions Viviane Pilla 1,2,*, Augusto C. Gonçalves 1,3, Alcindo A. Dos Santos 3 and Carlos Lodeiro 1,4 ID 1 BIOSCOPE Group, LAQV-REQUIMTE, Chemistry Department, Faculty of Science and Technology, University NOVA of Lisbon, 2829-516 Caparica, Portugal; [email protected] (A.C.G.); [email protected] (C.L.) 2 Instituto de Física, Universidade Federal de Uberlândia-UFU, Av. João Naves de Ávila 2121, 38400-902 Uberlândia, Brazil 3 Instituto de Química, Universidade de São Paulo-USP, Av. Prof. Lineu Prestes 748, CxP 26077, 05508-000 São Paulo, Brazil; [email protected] 4 Proteomass Scientific Society, Rua dos Inventores, Madan Park, 2829-516 Caparica, Portugal * Correspondence: [email protected] Received: 11 May 2018; Accepted: 28 June 2018; Published: 30 June 2018 Abstract: Fluorescein and its derivatives are yellowish-green emitting dyes with outstanding potential in advanced molecular probes for different applications. In this work, two fluorescent compounds containing fluorescein and an amino acid residue (compounds 1 and 2) were photophysically explored. Compounds 1 and 2 were previously synthesized via ester linkage between fluorescein ethyl ester and Boc-ser (TMS)-OH or Boc-cys(4-methyl benzyl)-OH. Studies on the time-resolved fluorescence lifetime and relative fluorescence quantum yield (φ) were performed for 1 and 2 diluted in acetonitrile, ethanol, dimethyl sulfoxide, and tetrahydrofuran solvents. The discussion considered the dielectric constants of the studied solvents (range between 7.5 and 47.2) and the photophysical parameters. -

Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌solomek,̌ †,‡ Christian G
Review pubs.acs.org/CR Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌Solomek,̌ †,‡ Christian G. Bochet,§ Aurelień Blanc,∥ Richard Givens,⊥ Marina Rubina,⊥ Vladimir Popik,# Alexey Kostikov,# and Jakob Wirz∇ † Department of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic ‡ Research Centre for Toxic Compounds in the Environment, Faculty of Science, Masaryk University, Kamenice 3, 625 00 Brno, Czech Republic § Department of Chemistry, University of Fribourg, Chemin du Museé 9, CH-1700 Fribourg, Switzerland ∥ Institut de Chimie, University of Strasbourg, 4 rue Blaise Pascal, 67000 Strasbourg, France ⊥ Department of Chemistry, University of Kansas, 1251 Wescoe Hall Drive, 5010 Malott Hall, Lawrence, Kansas 66045, United States # Department of Chemistry, University of Georgia, Athens, Georgia 30602, United States ∇ Department of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056 Basel, Switzerland 7.3. Arylsulfonyl Group 160 7.4. Ketones: 1,5- and 1,6-Hydrogen Abstraction 160 7.5. Carbanion-Mediated Groups 160 7.6. Sisyl and Other Silicon-Based Groups 161 7.7. 2-Hydroxycinnamyl Groups 161 7.8. α-Keto Amides, α,β-Unsaturated Anilides, CONTENTS and Methyl(phenyl)thiocarbamic Acid 162 7.9. Thiochromone S,S-Dioxide 162 1. Introduction 119 7.10. 2-Pyrrolidino-1,4-Benzoquinone Group 162 2. Arylcarbonylmethyl Groups 121 7.11. Triazine and Arylmethyleneimino Groups 162 2.1. Phenacyl and Other Related Arylcarbonyl- 7.12. Xanthene and Pyronin Groups 163 methyl Groups 121 o 7.13. Retro-Cycloaddition Reactions 163 2.2. -Alkylphenacyl Groups 123 8. Sensitized Release 163 2.3. p-Hydroxyphenacyl Groups 125 p 8.1. -

Fluorescence Quantum Yield of Rhodamine 6G Using Pulsed Photoacoustic Technique
Pram~na - J. Phys., Vol. 34, No. 6, June 1990, pp. 585-590. © Printed in India. Fluorescence quantum yield of rhodamine 6G using pulsed photoacoustic technique P SATHY, REJI PHILIP, V P N NAMPOORI and C P G VALLABHAN Laser Division, Department of Physics, Cochin University of Science and Technology, Cochin 682 022, India MS received 29 July 1989; revised 31 March 1990 Abstract. Experimental method for measuring photoacoustic(PA) signals generated by a pulsed laser beam in liquids is described. The pulsed PA technique is found to be a convenient and accurate method for determination of quantum yield in fluorescent dye solutions. Concentration dependence of quantum yield of rhodamine 6G in water is studied using the above method. The results indicate that the quantum yield decreases with increase in concentration in the quenching region in agreement with. the existing reports based on radiometric measurements. Keywords. Rhodamine 6G; pulsed photoacoustics; fluorescence; quantum yield. PACS Nos 33-50; 33.10 The knowledge of fluorescence quantum efficiency of organic dyes is important in selecting efficient dye laser media. Even after the various corrections for system geometry, re-absorption, polarization etc., the accuracy of the quantum yield values obtained from photometric measurements is rather poor (Demas and Gosby 1971). In order to evaluate the absolute quantum efficiency we have to consider both the radiative and non-radiative relaxation processes taking place in the medium. As the contribution due to non-radiative processes is not directly measurable using the traditional optical detection methods, photoacoustic(PA) and thermal lens techniques have been adopted recently for this purpose. -

1 Basic Principles of Fluorescence Spectroscopy
j1 1 Basic Principles of Fluorescence Spectroscopy 1.1 Absorption and Emission of Light As fluorophores play the central role in fluorescence spectroscopy and imaging we will start with an investigation of their manifold interactions with light. A fluorophore is a component that causes a molecule to absorb energy of a specific wavelength and then re-remit energy at a different but equally specific wavelength. The amount and wavelength of the emitted energy depend on both the fluorophore and the chemical environment of the fluorophore. Fluorophores are also denoted as chromophores, historically speaking the part or moiety of a molecule responsible for its color. In addition, the denotation chromophore implies that the molecule absorbs light while fluorophore means that the molecule, likewise,emits light. The umbrella term used in light emission is luminescence, whereas fluorescence denotes allowed transitions with a lifetime in the nanosecond range from higher to lower excited singlet states of molecules. In the following we will try to understand why some compounds are colored and others are not. Therefore, we will take a closer look at the relationship of conjugation to color with fluorescence emission, and investigate the absorption of light at different wavelengths in and near the visible part of the spectrum of various compounds. For example, organic compounds (i.e., hydrocarbons and derivatives) without double or triple bonds absorb light at wavelengths below 160 nm, corre- À sponding to a photon energy of >180 kcal mol 1 (1 cal ¼ 4.184 J), or >7.8 eV (Figure 1.1), that is, significantly higher than the dissociation energy of common carbon-to-carbon single bonds. -

Photochemical Production of Hydrogen Peroxide and Methylhydroperoxide in Coastal Waters
Marine Chemistry 97 (2005) 14–33 www.elsevier.com/locate/marchem Photochemical production of hydrogen peroxide and methylhydroperoxide in coastal waters Daniel W. O’Sullivan a,*, Patrick J. Neale b, Richard B. Coffin c, Thomas J. Boyd c, Christopher L. Osburn c a United States Naval Academy, Chemistry Department, 572 Holloway Road, Annapolis, MD 21402, USA b Smithsonian Environmental Research Center, 647 Contees Wharf Road, Edgewater, MD 21037, USA c Marine Biogeochemistry Section, Code 6114, US Naval Research Laboratory, Washington, DC 20375, USA Received 27 April 2004; received in revised form 5 April 2005; accepted 19 April 2005 Available online 13 September 2005 Abstract Hydrogen peroxide (H2O2) has been observed in significant concentrations in many natural waters. Because hydrogen peroxide can act as an oxidant and reductant, it participates in an extensive suite of reactions in surface waters. Hydrogen peroxide is produced as a secondary photochemical product of chromophoric dissolved organic matter (CDOM) photolysis. Apparent quantum yields for the photochemical production of hydrogen peroxide were determined in laboratory irradiations of filtered surface waters from several locations in the Chesapeake Bay and in Arctic coastal waters with varying levels of CDOM. The apparent quantum yield for H2O2 decreases by about an order of magnitude from 280 nm to 500 nm, and the majority of H2O2 production occurs at wavelengths less than 340 nm. The apparent quantum yield for H2O2 production at 290 nm ranged À 4 À 6 À 1 from 4.2Â10 to 2.1Â10 mol H2O2 (mol photons) from freshwater to marine waters. A linear relationship was found between the production of H2O2 and change in CDOM absorbance characterized as photobleaching (loss of absorbance). -
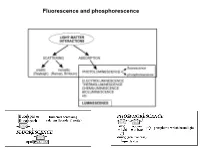
Fluorescence and Phosphorescence Possible De-Excitation Pathways of Excited Molecules
Fluorescence and phosphorescence Possible de-excitation pathways of excited molecules. Various parameters influence the emission of fluorescence. Luminescence: emission of photons from electronically excited states of atoms, molecules and ions. Fluorescence: a process in which a part of energy (UV, Visible) absorbed by a substance is released in the form of light as long as the stimulating radiation is continued. The fluorescence emission took place from a singlet excited states (average lifetime: from <10-10 to 10-7 sec). Phosphorescence: a process in which energy of light absorbed by a substance is released relatively slowly in the form of light. The phosphorescence emission took place from a triplet excited states (average lifetime: from 10-5 to >10+3 sec). S0: fundamental electronic state The Perrin–Jablonski diagram S0, S1, S2: singlet electronic states T1, T2: triplet electronic states Possible processes: ► photon absorption ► vibrational relaxation ► internal conversion ► intersystem crossing ► fluorescence ► phosphorescence ► delayed fluorescence ► triplet–triplet transitions Caracteristic times: absorption: 10-15 s vibrational relaxation: 10-12-10-10 s internal conversion: 10-11-10-9 s intersystem crossing: 10-10-10-8 s lifetime of the excited states: 10-10-10-7 s (fluorescence) lifetime of the excited states:10-6 s (phosphorescence) The vertical arrows corresponding to absorption start from the 0 (lowest) vibrational energy level of S0 because the majority of molecules are in this level at room temperature. Absorption of a photon can bring a molecule to one of the vibrational levels of S1; S2; . : For some aromatic hydrocarbons (naphthalene, anthracene and perylene) the absorption and fluorescence spectra exhibit vibrational bands.