Molecular Rearrangements of Superelectrophiles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Is There an Acid Strong Enough to Dissolve Glass? – Superacids
ARTICLE Is there an acid strong enough to dissolve glass? – Superacids For anybody who watched cartoons growing up, the word unit is based on how acids behave in water, however as acid probably springs to mind images of gaping holes being very strong acids react extremely violently in water this burnt into the floor by a spill, and liquid that would dissolve scale cannot be used for the pure ‘common’ acids (nitric, anything you drop into it. The reality of the acids you hydrochloric and sulphuric) or anything stronger than them. encounter in schools, and most undergrad university Instead, a different unit, the Hammett acidity function (H0), courses is somewhat underwhelming – sure they will react is often preferred when discussing superacids. with chemicals, but, if handled safely, where’s the drama? A superacid can be defined as any compound with an People don’t realise that these extraordinarily strong acids acidity greater than 100% pure sulphuric acid, which has a do exist, they’re just rarely seen outside of research labs Hammett acidity function (H0) of −12 [1]. Modern definitions due to their extreme potency. These acids are capable of define a superacid as a medium in which the chemical dissolving almost anything – wax, rocks, metals (even potential of protons is higher than it is in pure sulphuric acid platinum), and yes, even glass. [2]. Considering that pure sulphuric acid is highly corrosive, you can be certain that anything more acidic than that is What are Superacids? going to be powerful. What are superacids? Its all in the name – super acids are intensely strong acids. -

The Strongest Acid Christopher A
Chemistry in New Zealand October 2011 The Strongest Acid Christopher A. Reed Department of Chemistry, University of California, Riverside, California 92521, USA Article (e-mail: [email protected]) About the Author Chris Reed was born a kiwi to English parents in Auckland in 1947. He attended Dilworth School from 1956 to 1964 where his interest in chemistry was un- doubtedly stimulated by being entrusted with a key to the high school chemical stockroom. Nighttime experiments with white phosphorus led to the Headmaster administering six of the best. He obtained his BSc (1967), MSc (1st Class Hons., 1968) and PhD (1971) from The University of Auckland, doing thesis research on iridium organotransition metal chemistry with Professor Warren R. Roper FRS. This was followed by two years of postdoctoral study at Stanford Univer- sity with Professor James P. Collman working on picket fence porphyrin models for haemoglobin. In 1973 he joined the faculty of the University of Southern California, becoming Professor in 1979. After 25 years at USC, he moved to his present position of Distinguished Professor of Chemistry at UC-Riverside to build the Centre for s and p Block Chemistry. His present research interests focus on weakly coordinating anions, weakly coordinated ligands, acids, si- lylium ion chemistry, cationic catalysis and reactive cations across the periodic table. His earlier work included extensive studies in metalloporphyrin chemistry, models for dioxygen-binding copper proteins, spin-spin coupling phenomena including paramagnetic metal to ligand radical coupling, a Magnetochemi- cal alternative to the Spectrochemical Series, fullerene redox chemistry, fullerene-porphyrin supramolecular chemistry and metal-organic framework solids (MOFs). -

Superacid Chemistry
SUPERACID CHEMISTRY SECOND EDITION George A. Olah G. K. Surya Prakash Arpad Molnar Jean Sommer SUPERACID CHEMISTRY SUPERACID CHEMISTRY SECOND EDITION George A. Olah G. K. Surya Prakash Arpad Molnar Jean Sommer Copyright # 2009 by John Wiley & Sons, Inc. All rights reserved Published by John Wiley & Sons, Inc., Hoboken, New Jersey Published simultaneously in Canada No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 750-4470, or on the web at www.copyright.com. Requests to the Publisher for permission should be addressed to the Permissions Department, John Wiley & Sons, Inc., 111 River Street, Hoboken, NJ 07030, (201) 748-6011, fax (201) 748-6008, or online at http://www.wiley.com/go/permission. Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created or extended by sales representatives or written sales materials. The advice and strategies contained herein may not be suitable for your situation. -

George A. Olah 151
MY SEARCH FOR CARBOCATIONS AND THEIR ROLE IN CHEMISTRY Nobel Lecture, December 8, 1994 by G EORGE A. O L A H Loker Hydrocarbon Research Institute and Department of Chemistry, University of Southern California, Los Angeles, CA 90089-1661, USA “Every generation of scientific men (i.e. scientists) starts where the previous generation left off; and the most advanced discov- eries of one age constitute elementary axioms of the next. - - - Aldous Huxley INTRODUCTION Hydrocarbons are compounds of the elements carbon and hydrogen. They make up natural gas and oil and thus are essential for our modern life. Burning of hydrocarbons is used to generate energy in our power plants and heat our homes. Derived gasoline and diesel oil propel our cars, trucks, air- planes. Hydrocarbons are also the feed-stock for practically every man-made material from plastics to pharmaceuticals. What nature is giving us needs, however, to be processed and modified. We will eventually also need to make hydrocarbons ourselves, as our natural resources are depleted. Many of the used processes are acid catalyzed involving chemical reactions proceeding through positive ion intermediates. Consequently, the knowledge of these intermediates and their chemistry is of substantial significance both as fun- damental, as well as practical science. Carbocations are the positive ions of carbon compounds. It was in 1901 that Norris la and Kehrman lb independently discovered that colorless triphe- nylmethyl alcohol gave deep yellow solutions in concentrated sulfuric acid. Triphenylmethyl chloride similarly formed orange complexes with alumi- num and tin chlorides. von Baeyer (Nobel Prize, 1905) should be credited for having recognized in 1902 the salt like character of the compounds for- med (equation 1). -

Isolation and Characterization of a Non-Rigid Hexamethylbenzene-SO 2+ Complex Moritz Malischewski, Konrad Seppelt
Isolation and Characterization of a Non-Rigid Hexamethylbenzene-SO 2+ Complex Moritz Malischewski, Konrad Seppelt To cite this version: Moritz Malischewski, Konrad Seppelt. Isolation and Characterization of a Non-Rigid Hexamethylbenzene-SO 2+ Complex. Angewandte Chemie International Edition, Wiley-VCH Verlag, 2017, 56 (52), pp.16495-16497. 10.1002/anie.201708552. hal-01730776 HAL Id: hal-01730776 https://hal.sorbonne-universite.fr/hal-01730776 Submitted on 13 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Isolation and characterization of a non-rigid hexamethylbenzene- SO2+ complex Moritz Malischewski*[a],[b] and Konrad Seppelt[a] In memoriam of George Olah (1927-2017) Abstract: During our preparation of the pentagonal-pyramidal up besides recrystallization directly from the reaction mixture is 2+ 2+ - hexamethylbenzene-dication C6(CH3)6 we isolated the possible. Although C6(CH3)6SO (AsF6 )2 is the main component 2+ - unprecedented dicationic species C6(CH3)6SO (AsF6 )2 from the of the isolated material, ionic side-products co-precipitate due to reaction of hexamethylbenzene with a mixture of anhydrous HF, their low solubility in HF at low temperatures. AsF5 and liquid SO2. This compound can be understood as a complex of unknown SO2+ with hexamethylbenzene. -

Abstract Superacid Catalyzed Reactions
ABSTRACT SUPERACID CATALYZED REACTIONS: GENERATION OF REACTIVE INTERMEDIATES AND THEIR CHEMISTRY Makafui Gasonoo, Ph.D. Department of Chemistry and Biochemistry Northern Illinois University, 2017 Douglas A. Klumpp, Director This dissertation describes the use of triflic acid as catalyst for generating reactive intermediates and studying their reactivities under varying reaction conditions. The first chapter is an introduction to the types of organic reactions and acids. This chapter also discusses generation and reactivities of superelectrophiles as well. Chapter 2 discusses the synthesis of 3,3-disubstituted-2-oxindoles from the reaction of a series of acetonyl-substituted 3-hydroxy-2-oxindoles with arene in the presence of triflic acid. These reactions are performed under mild conditions and products isolated in decent yields. In chapter 3, the effects of charge migration in a tetra- and pentacationic superelectrophile is studied. These highly reactive intermediates are reacted with benzene at elevated temperatures. In the absence of benzene, cyclization occurs to produce novel N- heterocyclic compounds. Alcohol precursors are also ionized to generate these reactive intermediates and studied using low-temperature NMR. Chapter 4 talks about the effects of charge-charge repulsion on the aromaticity and anti- aromaticity in fluorenyl and dibenzosuberenyl cations respectively. These cations are generated from ionization of the respective biaryl ketones and dibenzosuberenols with superacid. With increasing charge, there is a corresponding increase in the aromaticity or anti-aromaticity of the respective system. Chapter 5 describes the superacid-promoted synthesis of heterocycle-containing 9,9- diarylfluorenes from biaryl ketones. Products are generally isolated in good yields using mild reaction conditions. Some of these compounds are used in the manufacture of organic light emitting diodes (OLEDs) and other organic-based electronics. -

George Andrew Olah Across Conventional Lines
GENERAL ARTICLE George Andrew Olah Across Conventional Lines Ripudaman Malhotra, Thomas Mathew and G K Surya Prakash Hungarian born American chemist, George Andrew Olah was aprolific researcher. The central theme of his career was the 12 pursuit of structure and mechanisms in chemistry, particu- larly focused on electron-deficient intermediates. He leaves behind a large body of work comprising almost 1500 papers and twenty books for the scientific community. Some selected works have been published in three aptly entitled volumes, 3 Across Conventional Lines. There is no way to capture the many contributions of Olah in a short essay. For this appreciation, we have chosen to high- 1 light some of those contributions that to our mind represent Ripudaman Malhotra, a student of Prof. Olah, is a asignificant advance to the state of knowledge. retired chemist who spent his entire career at SRI International working mostly Organofluorine Chemistry on energy-related issues. He co-authored the book on Early on in his career, while still in Hungary, Olah began studying global energy – A Cubic Mile organofluorine compounds. Olah’s interest in fluorinated com- of Oil. pounds was piqued by the theoretical ramifications of a strong 2 Thomas Mathew, a senior C-F bond. Thus, whereas chloromethanol immediately decom- scientist at the Loker Hydrocarbon Research poses into formaldehyde and HCl, he reasoned that the stronger Institute, has been a close C-F bond might render fluoromethanol stable. He succeeded in associate of Prof. Olah over preparing fluoromethanol by the reduction of ethyl flouorofor- two decades. 3 mate with lithium aluminum hydride [1]. -

Finding the Effects of Acids on Carbonates
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by TED Ankara College IB Thesis TED ANKARA COLLEGE FOUNDATION HIGH SCHOOL CHEMISTRY Finding the Effects of Acids on Carbonates Supervisor: Sedef Eryurt Candidate name, surname: Gülce Sözen 2010 1 Abstract Around the world, it is observed that lots of buildings, historical places are harmed. The damaged buildings have in common the features of the rainfall in their places. Effect of acid rain in these types of locations had been observed. Damages of the buildings have emerged because of the acid rains. The purpose of this project is observing the effects of different acids on the material of these types of buildings. The experimental design was prepared with the acids which we have got high probability to experience in our daily lives and with the building materials that can be characterized as carbonates. And also as carbonates, marbles has been preferred since they are more similar to the structures of buildings, where the carbonate as the dense. Used acids can be sorted into coke, vinegar, lemon juice, nitric acid and sulfuric acid. By comparing the observations, the most effective acid on carbonates and also the most powerful acid in the combination of acid rain can be found. This extended essay contains the background information about the subject, the collected data and analysis of these data. In addition, it includes the prevention and protection ways for the buildings and any other materials that can be affected by acids. (231 words) 2 TABLE OF CONTENTS 1. -

THÈSE De DOCTORAT Présentée Par Vuk VUKOVIĆ
UNIVERSITÉ DE STRASBOURG ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES (ED 222) Laboratoire de Catalyse Chimique Institut de Science et d'Ingénierie Supramoléculaires (UMR 7006) THÈSE de DOCTORAT présentée par Vuk VUKOVIĆ soutenue le 14 décembre 2018 pour obtenir le grade de : Docteur de l’Université de Strasbourg Discipline/ Spécialité : Chimie organique Synergistic Effect of Acids and HFIP on Friedel-Crafts Reactions of Alcohols and Cyclopropanes THESE dirigée par : Prof. MORAN Joseph Université de Strasbourg RAPPORTEURS : Prof. THIBAUDEAU Sébastien Université de Poitiers Prof. GANDON Vincent Université Paris-Sud 11 EXAMINATEUR: Dr WENCEL-DELORD Joanna Université de Strasbourg Synergistic Effect of Acids and HFIP on Friedel-Crafts Reactions of Alcohols and Cyclopropanes Table of Contents page Table of Contents I Acknowledgement and Dedication V List of Abbreviations XIII Résumé XVII Chapter 1. Genral introduction 1 1. 1. General aspects of Friedel-Crafts reactions 2 1. 1. 1. Substrates in Friedel-Crafts reactions 3 1. 2. Dehydrative functionalizations of alcohols 3 1. 2. 1. Substitution of alcohols via an SN2 pathway 4 1. 2. 2. Direct substitution of alcohols via an SN1 pathway 5 1. 2. 3. Distinguishing an SN1 from an SN2 process 6 1. 2. 4. Carbocationic intermediates and their reactivity 9 1. 2. 5. The “ease” of carbocation formation 12 1. 2. 6. Functionalization of alcohols via a hydrogen borrowing strategy 13 1. 2. 7. Catalysts for direct dehydroarylative substitution of alcohols 13 1. 3. HFIP as solvent and co-solvent in organic chemistry 14 1. 3. 1. Carbocations in HFIP 16 1. 3. 2. Self-association in HFIP 19 1. -

Isolating Benzenium Ion Salts
Published on Web 01/22/2003 Isolating Benzenium Ion Salts Christopher A. Reed,*,† Kee-Chan Kim,† Evgenii S. Stoyanov,‡ Daniel Stasko,† Fook S. Tham,† Leonard J. Mueller,† and Peter D. W. Boyd§ Contribution from the Department of Chemistry, UniVersity of California, RiVerside, California 92521-0403, Department of Chemistry, The UniVersity of Auckland, PriVate Bag, Auckland, New Zealand, and BoreskoV Institute of Catalysis, Prospekt LaVrentieVa, 5, NoVosibirsk, 630090, Russia Received June 17, 2002 ; E-mail: [email protected] Abstract: When partnered with carborane anions, arenium ions are remarkably stable. Previously investigated only at subambient temperatures in highly superacidic media, protonated benzene is readily isolated as a crystalline salt, thermally stable to >150 °C. Salts of the type [H(arene)][carborane] have been prepared by protonating benzene, toluene, m-xylene, mesitylene, and hexamethylbenzene with the carborane superacid H(CB11HR5X6)(R) H, Me; X ) Cl, Br). They have been characterized by elemental analysis, X-ray crystallography, NMR and IR methods. Solid-state 13C NMR spectra are similar to those observed earlier in solution, indicating that lattice interactions are comparable to solution solvation effects. The acidic proton(s) of the arenium cations interact weakly with the halide substituents of the anion via ion pairing. This is reflected in the dependence of the C-H stretching frequency on the basicity of the carborane anion. Bond lengths in the arenium ions are consistent with predominant cyclohexadienyl cation character, but charge distribution within the cation is less well represented by this resonance form. Structural and + vibrational comparison to theory is made for the benzenium ion (C6H7 ) with density functional theory at B3LYP/6-31G* and B3P86/6-311+G(d,p) levels. -
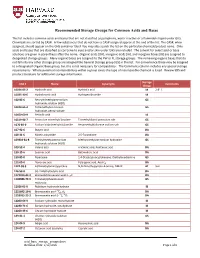
Common Acid and Base Storage Group Recommendations
Recommended Storage Groups for Common Acids and Bases This list includes common acids and bases that are not classified as pyrophoric, water reactive or a flammable liquid under GHS. Chemicals are sorted by CAS#. A few substances that do not have a CAS# assigned appear at the end of the list. The CAS#, when assigned, should appear on the GHS container label. You may also search the list on the particular chemical/product name. Only acids and bases that are classified as corrosive to eyes and/or skin under GHS are included. The solvent for select acid or base solutions are given in parenthesis after the name. Organic acids [OA], inorganic acids [IA], and inorganic bases [IB] are assigned to designated storage groups. Many organic bases are assigned to the PW or FL storage groups. The remaining organic bases that do not fall into any other storage group are assigned the General Storage group [GS] in this list. For convenience these may be assigned to a designated Organic Base group, but this is not necessary for compatibility. The Comments column includes any special storage requirements. Where potential incompatibilities within a group exists the type of incompatible chemical is listed. Review SDS and product literature for additional storage information. Storage CAS # Name Synonyms Comments Group 10034-85-2 Hydriodic acid Hydriotic acid IA 2-8° C 10035-10-6 Hydrobromic acid Hydrogen Bromide IA 100-85-6 Benzyltrimethylammonium GS hydroxide solution (H2O) 10424-65-4 Tetramethylammonium GS hydroxide, pentahydrate 10450-60-9 Periodic acid -
![The Strongest Brønsted Acid. Protonation of Alkanes at Room Temp.[**]](https://docslib.b-cdn.net/cover/1770/the-strongest-br%C3%B8nsted-acid-protonation-of-alkanes-at-room-temp-5391770.webp)
The Strongest Brønsted Acid. Protonation of Alkanes at Room Temp.[**]
What’s eating you, DOI: 10.1002/anie.200((will be filled in by the editorial staff)) alkane? The Strongest Brønsted acid. Protonation of Alkanes at Room Temp.[**] Matthew Nava, Irina V. Stoyanova, Steven Cummings, Evgenii S. Stoyanov, and Christopher A. Reed* For the past decade, the strongest acid to be isolated and fully considerably stronger acid than H(CHB11Cl11), capable of reacting characterized has been the chlorinated carborane acid, rapidly with all organics, and being the ultimate desiccant. So, [1,2] H(CHB11Cl11). More recently, this has been equaled by the purity of starting materials and the scrupulous exclusion of water comparably strong, isoelectronic, all-boron diprotic superacid, were imperative. In particular, we found that the trityl salt of [3] − H2(B12Cl12). The superior strength of these Brønsted acids has CHB11F11 must be carefully purified by successive been established in all phases. In solution, the fact that both readily recrystallizations from o-dichlorobenzene in order to remove protonate benzene whereas oxyacids do not, places them ahead of occluded organics. Another critical step was to perform the reaction [4] HFSO3, the previous strongest pure acid measured on the Ho scale. of [Et3Si-H-SiEt3][CHB11F11] with HCl twice (sequentially) in order In the gas phase, the calculated[5] and measured[6] deprotonation to completely remove silane byproducts. Once isolated, small enthalpy of H(CHB11Cl11) is the lowest of any available acid. The quantities of H(CHB11F11) were obtained in high purity by νNH anion basicity scale[7] indicates that carborane anions are less sublimation under high vacuum at ca. 160°C, a temperature similar basic than the oxyanions of traditional acids.