Table of Thermal Properties of Linear Macromolecules and Related Small Molecules—The ATHAS Data Bank A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Feas: Heat Capacity, Enthalpy Increments, Other Thermodynamic Properties from 5 to 1350 K, and Magnetic Transition A
M-2341 J. Chem. Thermodynumics 1989,21, 363-373 FeAs: Heat capacity, enthalpy increments, other thermodynamic properties from 5 to 1350 K, and magnetic transition a DOMINGO GONZALEZ-ALVAREZ, Fact&ad de Ciencias de la Universidad de Zaragoza, Departamento de Fisica Fundamental, 5009 Zaragoza, Spain FREDRIK GR0NVOLD, Chemical Institute, University of Oslo, Blindern, Oslo 3, Norway BENGT FALK,b EDGAR F. WESTRUM, JR., Department of Chemistry, University of Michigan, Ann Arbor, MI 48109, U.S.A. R. BLACHNIK,’ and G. KUDERMANNd Technical University, Siegen, Federal Republic of Germany (Received 2 January 1989) The heat capacity of iron monoarsenide has been determined by adiabatic calorimetry from 5 to 1030 K and bv drou calorimetrv relative to 298.15 K over the range 875 to 1350 K. A small h-type transition is observed at ?;, = (70.95 kO.02) K. It is related & the disappearance of a doubly helically ordered magnetic-spin structure on heating. The obviously cooperative entropy increment of transition is only A&JR = 0.021. The higher-temperature heat capacity rises considerably above lattice expectations. Part of the rise is ascribed to low-spin electron redistribution in iron, while the further excess above 800 K presumably arises from a beginning low- to high-spin transition, possibly connected with interstitial defect formation in the MnP- type structure. FeAs melts at about 1325 K with A,,,Hk=6180R.K. Thermodynamic functions have been evaluated and the values of C,,,(T), S:(T), H:(T), and @i(T), are 6.057R, 7.513R,1177R.K, and 3.567Rat 298.15 K, and 8.75R,16.03R, 6287R.K, and 9.74512at 1000 K. -

High Temperature Enthalpies of the Lead Halides
HIGH TEMPERATURE ENTHALPIES OF THE LEAD HALIDES: ENTHALPIES AND ENTROPIES OF FUSION APPROVED: Graduate Committee: Major Professor Committee Member Min fessor Committee Member X Committee Member UJ. Committee Member Committee Member Chairman of the Department of Chemistry Dean or the Graduate School HIGH TEMPERATURE ENTHALPIES OF THE LEAD HALIDES: ENTHALPIES AND ENTROPIES OF FUSION DISSERTATION Presented to the Graduate Council of the North Texas State University in Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY By Clarence W, Linsey, B. S., M. S, Denton, Texas June, 1970 ACKNOWLEDGEMENT This dissertation is based on research conducted at the Oak Ridge National Laboratory, Oak Ridge, Tennessee, which is operated by the Uniofl Carbide Corporation for the U. S. Atomic Energy Commission. The author is also indebted to the Oak Ridge Associated Universities for the Oak Ridge Graduate Fellowship which he held during the laboratory phase. 11 TABLE OF CONTENTS Page LIST OF TABLES iv LIST OF ILLUSTRATIONS v Chapter I. INTRODUCTION . 1 Lead Fluoride Lead Chloride Lead Bromide Lead Iodide II. EXPERIMENTAL 11 Reagents Fusion-Filtration Method Encapsulation of Samples Equipment and Procedure Bunsen Ice Calorimeter Computer Programs Shomate Method Treatment of Data Near the Melting Point Solution Calorimeter Heat of Solution Calculations III. RESULTS AND DISCUSSION 41 Lead Fluoride Variation in Heat Contents of the Nichrome V Capsules Lead Chloride Lead Bromide Lead Iodide Summary BIBLIOGRAPHY ...... 98 in LIST OF TABLES Table Pa8e I. High-Temperature Enthalpy Data for Cubic PbE2 Encapsulated in Gold 42 II. High-Temperature Enthalpy Data for Cubic PbF2 Encapsulated in Molybdenum 48 III. -

Molecular Dynamics Simulation Studies of Physico of Liquid
MD Simulation of Liquid Pentane Isomers Bull. Korean Chem. Soc. 1999, Vol. 20, No. 8 897 Molecular Dynamics Simulation Studies of Physico Chemical Properties of Liquid Pentane Isomers Seng Kue Lee and Song Hi Lee* Department of Chemistry, Kyungsung University, Pusan 608-736, Korea Received January 15, 1999 We have presented the thermodynamic, structural and dynamic properties of liquid pentane isomers - normal pentane, isopentane, and neopentane - using an expanded collapsed atomic model. The thermodynamic prop erties show that the intermolecular interactions become weaker as the molecular shape becomes more nearly spherical and the surface area decreases with branching. The structural properties are well predicted from the site-site radial, the average end-to-end distance, and the root-mean-squared radius of gyration distribution func tions. The dynamic properties are obtained from the time correlation functions - the mean square displacement (MSD), the velocity auto-correlation (VAC), the cosine (CAC), the stress (SAC), the pressure (PAC), and the heat flux auto-correlation (HFAC) functions - of liquid pentane isomers. Two self-diffusion coefficients of liq uid pentane isomers calculated from the MSD's via the Einstein equation and the VAC's via the Green-Kubo relation show the same trend but do not coincide with the branching effect on self-diffusion. The rotational re laxation time of liquid pentane isomers obtained from the CAC's decreases monotonously as branching increas es. Two kinds of viscosities of liquid pentane isomers calculated from the SAC and PAC functions via the Green-Kubo relation have the same trend compared with the experimental results. The thermal conductivity calculated from the HFAC increases as branching increases. -

Functionalized Aromatics Aligned with the Three Cartesian Axes: Extension of Centropolyindane Chemistry*
Pure Appl. Chem., Vol. 78, No. 4, pp. 749–775, 2006. doi:10.1351/pac200678040749 © 2006 IUPAC Functionalized aromatics aligned with the three Cartesian axes: Extension of centropolyindane chemistry* Dietmar Kuck‡ Fakultät für Chemie, Universität Bielefeld, Universitätsstraße 25, D-33615 Bielefeld, Germany Abstract: The unique geometrical features and structural potential of the centropolyindanes, a complete family of novel, 3D polycyclic aromatic hydrocarbons, are discussed with respect to the inherent orthogonality of their arene units. Thus, the largest member of the family, centrohexaindane, a topologically nonplanar hydrocarbon, is presented as a “Cartesian hexa- benzene”, because each of its six benzene units is stretched into one of the six directions of the Cartesian space. This feature is discussed on the basis of the X-ray crystal structures of centrohexaindane and two lower members of the centropolyindane family, viz. the parent tribenzotriquinacenes. Recent progress in multiple functionalization and extension of the in- dane wings of selected centropolyindanes is reported, including several highly efficient six- and eight-fold C–C cross-coupling reactions. Some particular centropolyindane derivatives are presented, such as the first twelve-fold functionalized centrohexaindane and a tribenzo- triquinacene bearing three mutually orthogonal phenanthroline groupings at its molecular pe- riphery. Challenges to further extend the arene peripheries of the tribenzotriquinacenes and fenestrindanes to give, eventually, graphite cuttings bearing a central bowl- or saddle-shaped center are outlined, as is the hypothetical generation of a “giant” nanocube consisting of eight covalently bound tribenzotriquinacene units. Along these lines, our recent discovery of a re- lated, solid-state supramolecular cube, containing eight molecules of a particular tri- bromotrinitrotribenzotriquinacene of the same absolute configuration, is presented for the first time. -

THERMODYNAMIC EVALUATION of the Cu-Ti SYSTEM in VIEW of SOLID STATE AMORPHIZATION REACTIONS L
THERMODYNAMIC EVALUATION OF THE Cu-Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS L. Battezzati, M. Baricco, G. Riontino, I. Soletta To cite this version: L. Battezzati, M. Baricco, G. Riontino, I. Soletta. THERMODYNAMIC EVALUATION OF THE Cu- Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS. Journal de Physique Colloques, 1990, 51 (C4), pp.C4-79-C4-85. 10.1051/jphyscol:1990409. jpa-00230769 HAL Id: jpa-00230769 https://hal.archives-ouvertes.fr/jpa-00230769 Submitted on 1 Jan 1990 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ~OLLOQUEDE PHYSIQUE Colloque C4, suppl6ment au 11'14, Tome 51, 15 juillet 1990 THERMODYNAMIC EVALUATION OF THE Cu-Ti SYSTEM IN VIEW OF SOLID STATE AMORPHIZATION REACTIONS L. BATTEZZATI* , M. BARICCO*~ *""" , G. RIONTINO" *"* and I. SOLETTA** * '~ipartimento di Chimica Inorganics, Chimica Fisica e Chimica dei Materiali, Universitd di Torino, Italy ""~stitutoElettrotecnico Nazionale Galileo Perraris, Torino, Italy *.* Dipartimento di Chimica, Universitd di Sassari, Italy *et* INPM, Unita' di Ricerca di Torino, Italy Resumk -La description thermodynamique du sist6me Cu-Ti est reconsideree, car les calculs he la littgrature, bien que donnant une bonne representation du diagramme de phase, ne prevoient pas la possibilit6 de l'amorphisation. -

2.6 Physical Properties of Alkanes
02_BRCLoudon_pgs4-4.qxd 11/26/08 8:36 AM Page 70 70 CHAPTER 2 • ALKANES PROBLEMS 2.12 Represent each of the following compounds with a skeletal structure. (a) CH3 CH3CH2CH2CH" CH C(CH3)3 L L "CH3 (b) ethylcyclopentane 2.13 Name the following compounds. (a) (b) CH3 CH2CH3 H3C 2.14 How many hydrogens are in an alkane of n carbons containing (a) two rings? (b) three rings? (c) m rings? 2.15 How many rings does an alkane have if its formula is (a) C8H10? (b) C7H12? Explain how you know. 2.6 PHYSICAL PROPERTIES OF ALKANES Each time we come to a new family of organic compounds, we’ll consider the trends in their melting points, boiling points, densities, and solubilities, collectively referred to as their phys- ical properties. The physical properties of an organic compound are important because they determine the conditions under which the compound is handled and used. For example, the form in which a drug is manufactured and dispensed is affected by its physical properties. In commercial agriculture, ammonia (a gas at ordinary temperatures) and urea (a crystalline solid) are both very important sources of nitrogen, but their physical properties dictate that they are handled and dispensed in very different ways. Your goal should not be to memorize physical properties of individual compounds, but rather to learn to predict trends in how physical properties vary with structure. A. Boiling Points The boiling point is the temperature at which the vapor pressure of a substance equals atmos- pheric pressure (which is typically 760 mm Hg). -

The Influence of Electron Beam Irradiation on the Property Behaviour of Medical Grade Poly (Ether-Block-Amide) (PEBA)
Australian Journal of Basic and Applied Sciences, 7(5): 174-181, 2013 ISSN 1991-8178 The Influence Of Electron Beam Irradiation On The Property Behaviour Of Medical Grade Poly (Ether-Block-Amide) (PEBA) 1Kieran A. Murray, 1James E.Kennedy, 2Brian McEvoy, 2Olivier Vrain, 2Damien Ryan, 2Richard Cowman and 1Clement L. Higginbotham 1Materials Research Institute, Athlone Institute of Technology, Dublin Road, Athlone, Co. Westmeath, Ireland 2 Synergy Health Applied Sterilisation Technologies, IDA Business & Technology Park, Sragh, Tullamore, Co. Offaly, Ireland Abstract: The aim of this paper is to investigate the potential effects of electron beam, ranging between 5 and 200kGy for unstabilised Poly (ether-block-amide) (Pebax) material. The Pebax samples were subjected to a wide range of extensive characterisation techniques in order to identify any modifications to the material properties. The results demonstrated that a series of changes occurred and some of these included a reduction in the mechanical properties and melting strength, an increase in the molecular weight and viscosity and alterations to surface characteristics of the material. Overall, this study provided evidence that electron beam irradiation lead to the development of simultaneous crosslinking and chain scission in the Pebax material, where crosslinking/branching became the dominating factor above 10kGy. Key words: Electron beam irradiation,Poly(ether-block-amide), Thermal, mechanical, structural and surface properties, Temperature ramp, Degradation INTRODUCTION Poly (ether-block-amide) (Pebax) is a copolymer which consists of linear chains of rigid polyamide blocks covalently linked to flexible polyether blocks via ester groups (Joseph and Flesher, 1986). The polyamide crystalline region provides the mechanical strength while the polyether amorphous region offers high permeability as a result of the high chain mobility of the ether linkage (Joseph and Flesher, 1986; Liu, 2008). -
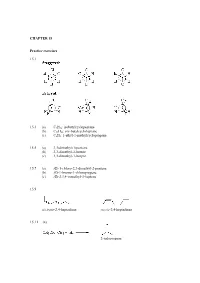
CHAPTER 15 Practice Exercises 15.1 15.3 (A) C9H18
CHAPTER 15 Practice exercises 15.1 15.3 (a) C9H18: isobutylcyclopentane (b) C11H22: sec-butylcycloheptane (c) C6H2: 1-ethyl-1-methylcyclopropane 15.5 (a) 3,3-dimethyl-1-pentene (b) 2,3-dimethyl-2-butene (c) 3,3-dimethyl-1-butyne 15.7 (a) (E)-1-chloro-2,3-dimethyl-2-pentene (b) (Z)-1-bromo-1-chloropropene (c) (E)-2,3,4-trimethyl-3-heptene 15.9 cis,trans-2,4-heptadiene cis,cis-2,4-heptadiene 15.11 (a) 2-iodopropane (b) 1-iodo-1-methylcyclohexane 15.13 CH3 CH3 + HI I Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step + CH3 HI CH3 I rate-determining step Step 2: Nucleophilic attack of the iodide anion on the 3˚ carbocation intermediate to give the product: CH + 3 CH3 I I 15.15 H2O CH3 CH3 + H3O OH Step 1: Protonation of the alkene to give the most stable 3˚ carbocation intermediate: slow step H + + CH3 HOH CH3 O H rate-determining H step Step 2: Nucleophilic attack of the water on the 3˚ carbocation intermediate to give the protonated alcohol: H H O+ H + CH3 O H CH3 Step 3: The protonated alcohol loses a proton to form the product: H + O H H CH3 + O + H3O CH3 H OH 15.17 (a) 2-phenyl-2-propanol (b) (E)-3,4-diphenyl-3-hexene (c) 3-methylbenzoic acid or m-methylbenzoic acid Review questions 15.1 A hydrocarbon is a compound composed only of hydrogen and carbon atoms. 15.3 In saturated hydrocarbons, each carbon is bonded to four other atoms, either hydrogen or carbon atoms. -

The Dynisco Extrusion Processors Handbook 2Nd Edition
The Dynisco Extrusion Processors Handbook 2nd edition Written by: John Goff and Tony Whelan Edited by: Don DeLaney Acknowledgements We would like to thank the following people for their contributions to this latest edition of the DYNISCO Extrusion Processors Handbook. First of all, we would like to thank John Goff and Tony Whelan who have contributed new material that has been included in this new addition of their original book. In addition, we would like to thank John Herrmann, Jim Reilly, and Joan DeCoste of the DYNISCO Companies and Christine Ronaghan and Gabor Nagy of Davis-Standard for their assistance in editing and publication. For the fig- ures included in this edition, we would like to acknowledge the contributions of Davis- Standard, Inc., Krupp Werner and Pfleiderer, Inc., The DYNISCO Companies, Dr. Harold Giles and Eileen Reilly. CONTENTS SECTION 1: INTRODUCTION TO EXTRUSION Single-Screw Extrusion . .1 Twin-Screw Extrusion . .3 Extrusion Processes . .6 Safety . .11 SECTION 2: MATERIALS AND THEIR FLOW PROPERTIES Polymers and Plastics . .15 Thermoplastic Materials . .19 Viscosity and Viscosity Terms . .25 Flow Properties Measurement . .28 Elastic Effects in Polymer Melts . .30 Die Swell . .30 Melt Fracture . .32 Sharkskin . .34 Frozen-In Orientation . .35 Draw Down . .36 SECTION 3: TESTING Testing and Standards . .37 Material Inspection . .40 Density and Dimensions . .42 Tensile Strength . .44 Flexural Properties . .46 Impact Strength . .47 Hardness and Softness . .48 Thermal Properties . .49 Flammability Testing . .57 Melt Flow Rate . .59 Melt Viscosity . .62 Measurement of Elastic Effects . .64 Chemical Resistance . .66 Electrical Properties . .66 Optical Properties . .68 Material Identification . .70 SECTION 4: THE SCREW AND BARREL SYSTEM Materials Handling . -

Efficient Approach to Compute Melting Properties Fully from Ab Initio With
PHYSICAL REVIEW B 96, 224202 (2017) Efficient approach to compute melting properties fully from ab initio with application to Cu Li-Fang Zhu,* Blazej Grabowski, and Jörg Neugebauer Max-Planck-Institut für Eisenforschung GmbH, D-40237 Düsseldorf, Germany (Received 3 August 2017; revised manuscript received 16 October 2017; published 13 December 2017) Applying thermodynamic integration within an ab initio-based free-energy approach is a state-of-the-art method to calculate melting points of materials. However, the high computational cost and the reliance on a good reference system for calculating the liquid free energy have so far hindered a general application. To overcome these challenges, we propose the two-optimized references thermodynamic integration using Langevin dynamics (TOR-TILD) method in this work by extending the two-stage upsampled thermodynamic integration using Langevin dynamics (TU-TILD) method, which has been originally developed to obtain anharmonic free energies of solids, to the calculation of liquid free energies. The core idea of TOR-TILD is to fit two empirical potentials to the energies from density functional theory based molecular dynamics runs for the solid and the liquid phase and to use these potentials as reference systems for thermodynamic integration. Because the empirical potentials closely reproduce the ab initio system in the relevant part of the phase space the convergence of the thermodynamic integration is very rapid. Therefore, the proposed approach improves significantly the computational efficiency while preserving the required accuracy. As a test case, we apply TOR-TILD to fcc Cu computing not only the melting point but various other melting properties, such as the entropy and enthalpy of fusion and the volume change upon melting. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Adsorption of Nitrogen, Neopentane, N-Hexane, Benzene and Methanol for the Evaluation of Pore Sizes in Silica Grades of MCM-41
Microporous and Mesoporous Materials 47 >2001) 323±337 www.elsevier.com/locate/micromeso Adsorption of nitrogen, neopentane, n-hexane, benzene and methanol for the evaluation of pore sizes in silica grades of MCM-41 M.M.L. Ribeiro Carrott a,*, A.J.E. Candeias a, P.J.M. Carrott a, P.I. Ravikovitch b, A.V. Neimark b, A.D. Sequeira c a Department of Chemistry, University of Evora, Colegio Luõs Antonio Verney, Rua Roma~o Romalho 59, 7000-671 Evora, Portugal b Center for Modeling and Characterization of Nanoporous Materials, TRI/Princeton, 601 Prospect Avenue, Princeton, NJ 08542-0625, USA c Department of Physics, Nuclear and Technological Institute, 2686-953 Sacavem, Portugal Received 26 February 2001; received in revised form 7 June 2001; accepted 11 June 2001 Abstract Nitrogen, neopentane, n-hexane, benzene and methanol adsorption isotherms were determined on ®ve samples of silica grade MCM-41 with dierent pore sizes and a comparison of dierent methods for evaluating the pore size was carried out. With nitrogen we found a remarkably good agreement between the results obtained from the non-local density functional theory and geometric methods, with corresponding values obtained by the two methods diering by less than 0.05 nm. On the other hand, the results con®rm ®ndings seen before by other workers that, in order to obtain reliable values of pore radii by the hydraulic method, it is necessary to use a cross-sectional area of nitrogen in the monolayer smaller than the normally assumed value of 0.162 nm2. In addition, it was found that the eective pore volume obtained with the four organic adsorptives was almost constant while the value obtained from nitrogen ad- sorption data was always higher.