Molecular Dynamics Simulation Studies of Physico of Liquid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methane Hydrocarbon Compounds During Wintertime in Beijing
Atmos. Chem. Phys. Discuss., doi:10.5194/acp-2016-783, 2016 Manuscript under review for journal Atmos. Chem. Phys. Published: 16 December 2016 c Author(s) 2016. CC-BY 3.0 License. The levels, variation characteristics and sources of atmospheric non- methane hydrocarbon compounds during wintertime in Beijing, China Chengtang Liu1,3, Yujing Mu1,2 *, Junfeng Liu1,3, Chenglong Zhang1,3, Yuanyuan Zhang1,3, Pengfei 5 Liu1,3, Hongxing Zhang1,4 1Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China 2Center for Excellence in Regional Atmospheric Environment, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China 3University of Chinese Academy of Sciences, Beijing 100085, China 10 4Beijing Urban Ecosystem Research Station, Beijing, 100085, China Correspondence to: Yujing Mu ([email protected]) Abstract. Atmospheric non-methane hydrocarbon compounds (NMHCs) were measured at a sampling site in Beijing city from 15 December 2015 to 14 January 2016 to recognize their pollution levels, variation characteristics and sources. Fifty- 15 three NMHCs were quantified and the proportions of alkanes, alkenes, acetylene and aromatics to the total NMHCs were 49.8% ~ 55.8%, 21.5% ~ 24.7%, 13.5% ~ 15.9% and 9.3% ~ 10.7%, respectively. The variation trends of the NMHCs concentrations were basically identical and exhibited remarkable fluctuation, which were mainly ascribed to the variation of meteorological conditions, especially wind speed. The diurnal variations of NMHCs in clear days exhibited two peaks during the morning and evening rush hours, whereas the rush hours’ peaks diminished or even disappeared in the haze days, 20 implying that the relative contribution of the vehicular emission to atmospheric NMHCs depended on the pollution status. -

Isopentane Ipt
ISOPENTANE IPT CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: -70°F C.C. 7.1 Grades of Purity: Research: 99.99%; pure: Common Synonyms Watery liquid Colorless Gasoline-like odor (approx.) 99.4%; technical: 97% 2-Methylbutane 4.2 Flammable Limits in Air: 1.4%-8.3% 7.2 Storage Temperature: Ambient Floats on water. Flammable, irritating vapor is produced. Boiling point 4.3 Fire Extinguishing Agents: Dry 7.3 Inert Atmosphere: No requirement chemical, foam, or carbon dioxide is 82°F. 7.4 Venting: Open (flame arrester) or pressure- 4.4 Fire Extinguishing Agents Not to Be vacuum Evacuate. Used: Water may be ineffective 7.5 IMO Pollution Category: C Keep people away. 4.5 Special Hazards of Combustion Wear goggles and self-contained breathing apparatus. Products: Not pertinent 7.6 Ship Type: 3 Shut off ignition sources and call fire department. 4.6 Behavior in Fire: Highly volatile liquid. 7.7 Barge Hull Type: Currently not available Avoid contact with liquid and vapor. Vapors may explode when mixed with air. Stay upwind and use water spray to ``knock down'' vapor. Notify local health and pollution control agencies. 4.7 Auto Ignition Temperature: 800°F 8. HAZARD CLASSIFICATIONS 4.8 Electrical Hazards: Not pertinent 8.1 49 CFR Category: Flammable liquid FLAMMABLE. Fire 4.9 Burning Rate: 7.4 mm/min. 8.2 49 CFR Class: 3 Flashback along vapor trail may occur. 4.10 Adiabatic Flame Temperature: Currently 8.3 49 CFR Package Group: I Vapor may explode if ignited in an enclosed area. -

TCEQ Interoffice Memorandum
TCEQ Interoffice Memorandum To: Tony Walker Director, TCEQ Region 4, Dallas/Fort Worth Alyssa Taylor Special Assistant to the Regional Director, TCEQ Region 4, Dallas/Fort Worth From: Shannon Ethridge, M.S., D.A.B.T. Toxicology Division, Office of the Executive Director Date: Draft, 2014 Subject: Toxicological Evaluation of Results from an Ambient Air Sample for Volatile Organic Compounds Collected Downwind of the EagleRidge Energy, LLC - Woodland Estates West Unit (Latitude 32.595248, Longitude -97.160361) in Mansfield, Tarrant County, Texas Sample Collected on November 26, 2013, Request Number 1312003 (Lab Sample 1312003-001) Key Points • Reported concentrations of target volatile organic compounds (VOCs) were either not detected or were detected below levels of short-term health and/or welfare concern. Background On November 26, 2013, a Texas Commission on Environmental Quality (TCEQ) Region 4 air investigator collected a 30-minute canister sample (Lab Sample 1312003-001) downwind of the EagleRidge Energy, LLC - Woodland Estates West Unit (Latitude 32.595248, Longitude -97.160361) in Mansfield, Tarrant County, Texas. The sample was collected in response to a citizen complaint of a sore throat. The investigator did not experience an odor or health effects while sampling. Meteorological conditions measured at the site or nearest stationary ambient air monitoring site indicated that the ambient temperature was 44.3°F with a relative humidity of 63.6%, and winds were from the north (360°) at 7.5 to 9.5 miles per hour. The sampling site was less than 100 feet from the nearest possible emission source (tanks). The nearest location where the public could have access was approximately 301 to 500 feet from the possible emission source. -

Functionalized Aromatics Aligned with the Three Cartesian Axes: Extension of Centropolyindane Chemistry*
Pure Appl. Chem., Vol. 78, No. 4, pp. 749–775, 2006. doi:10.1351/pac200678040749 © 2006 IUPAC Functionalized aromatics aligned with the three Cartesian axes: Extension of centropolyindane chemistry* Dietmar Kuck‡ Fakultät für Chemie, Universität Bielefeld, Universitätsstraße 25, D-33615 Bielefeld, Germany Abstract: The unique geometrical features and structural potential of the centropolyindanes, a complete family of novel, 3D polycyclic aromatic hydrocarbons, are discussed with respect to the inherent orthogonality of their arene units. Thus, the largest member of the family, centrohexaindane, a topologically nonplanar hydrocarbon, is presented as a “Cartesian hexa- benzene”, because each of its six benzene units is stretched into one of the six directions of the Cartesian space. This feature is discussed on the basis of the X-ray crystal structures of centrohexaindane and two lower members of the centropolyindane family, viz. the parent tribenzotriquinacenes. Recent progress in multiple functionalization and extension of the in- dane wings of selected centropolyindanes is reported, including several highly efficient six- and eight-fold C–C cross-coupling reactions. Some particular centropolyindane derivatives are presented, such as the first twelve-fold functionalized centrohexaindane and a tribenzo- triquinacene bearing three mutually orthogonal phenanthroline groupings at its molecular pe- riphery. Challenges to further extend the arene peripheries of the tribenzotriquinacenes and fenestrindanes to give, eventually, graphite cuttings bearing a central bowl- or saddle-shaped center are outlined, as is the hypothetical generation of a “giant” nanocube consisting of eight covalently bound tribenzotriquinacene units. Along these lines, our recent discovery of a re- lated, solid-state supramolecular cube, containing eight molecules of a particular tri- bromotrinitrotribenzotriquinacene of the same absolute configuration, is presented for the first time. -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Flammable Gas Mixture: Ethane / Isobutane / Isopentane / N-Butane / N-Pentane / Nitrogen / Propane Section 1. Identification GHS product identifier : Flammable Gas Mixture: Ethane / Isobutane / Isopentane / N-Butane / N-Pentane / Nitrogen / Propane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 008109 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE GASES - Category 1 substance or mixture GASES UNDER PRESSURE - Compressed gas SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 AQUATIC HAZARD (LONG-TERM) - Category 3 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable gas. Contains gas under pressure; may explode if heated. May form explosive mixtures in Air. May displace oxygen and cause rapid suffocation. May cause drowsiness and dizziness. Harmful to aquatic life with long lasting effects. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Approach suspected leak area with caution. -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -

2.6 Physical Properties of Alkanes
02_BRCLoudon_pgs4-4.qxd 11/26/08 8:36 AM Page 70 70 CHAPTER 2 • ALKANES PROBLEMS 2.12 Represent each of the following compounds with a skeletal structure. (a) CH3 CH3CH2CH2CH" CH C(CH3)3 L L "CH3 (b) ethylcyclopentane 2.13 Name the following compounds. (a) (b) CH3 CH2CH3 H3C 2.14 How many hydrogens are in an alkane of n carbons containing (a) two rings? (b) three rings? (c) m rings? 2.15 How many rings does an alkane have if its formula is (a) C8H10? (b) C7H12? Explain how you know. 2.6 PHYSICAL PROPERTIES OF ALKANES Each time we come to a new family of organic compounds, we’ll consider the trends in their melting points, boiling points, densities, and solubilities, collectively referred to as their phys- ical properties. The physical properties of an organic compound are important because they determine the conditions under which the compound is handled and used. For example, the form in which a drug is manufactured and dispensed is affected by its physical properties. In commercial agriculture, ammonia (a gas at ordinary temperatures) and urea (a crystalline solid) are both very important sources of nitrogen, but their physical properties dictate that they are handled and dispensed in very different ways. Your goal should not be to memorize physical properties of individual compounds, but rather to learn to predict trends in how physical properties vary with structure. A. Boiling Points The boiling point is the temperature at which the vapor pressure of a substance equals atmos- pheric pressure (which is typically 760 mm Hg). -

Analysis of Impurities in Ethylene by ASTM D6159-97
Petrochemical Applications Analysis of Impurities in Ethylene by ASTM D6159-97 Ethylene is one of the highest volume chemicals produced in the world, with global production exceed- ing 100 million metric tons annually. Ethylene is primarily used in the manufacture of polyethylene, ethylene oxide, and ethylene dichloride, as well as many other lower volume products. Most of these production processes use various catalysts to improve product quality and process yield. Impurities in ethylene can damage the catalysts, resulting in significant replacement costs, reduced product quality, process downtime, and decreased yield. Ethylene is typically manufactured through the use of steam cracking. In this process, gaseous or light liquid hydrocarbons are combined with steam and heated to 750–950°C in a pyrolysis furnace. Numerous free radical reactions are initiated and larger hydrocarbons are converted (cracked) into smaller hydro- carbons. The high temperatures used in steam cracking promote the formation of unsaturated or olefinic compounds like ethylene. Ethylene feedstocks must be tested to ensure that only high purity ethylene is delivered for subsequent chemical processing. Testing typically follows ASTM D6159-97, a GC/FID method which employs a two-column configura- tion consisting of an alumina PLOT column with KCl deactivation (50m x 0.53mm ID) coupled to a methyl silicone column (30m x 0.53mm ID x 5.0µm df). Figure 1 Methane and ethane are well resolved in high purity ethylene samples. Peak List 1. methane 2. ethane 3. ethylene GC_PC01109 Column: Rt®-Alumina BOND/KCl, 50m, 0.53mm ID, 10.0µm (cat.# 19760) in series with Rtx®-1, 30m, 0.53mm ID, 5.0µm (cat.# 10179), connected using a Universal Press-Tight® Connector (cat.# 20401) Sample: ethylene Inj.: 1µL split, 60mL/min. -
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Supplementary Material (ESI) for Chemical Communications This Journal Is © the Royal Society of Chemistry 2012
Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Supplementary Material Guest-controlled Self-Sorting in Assemblies driven by the Hydrophobic Effect Haiying Gan, Bruce C. Gibb* Department of Chemistry University of New Orleans LA 70148 Email: [email protected] S1 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Table of Contents General p S3 1H-NMR spectra of 1 and 2, and mixture of hosts, in the absence of guests p S4 1H-NMR spectra of the complexes of 1.2 and hydrocarbons n-pentane through n-heptadecane p S5-S8 Example COSY and NOESY NMR data (for n-undecane hetero-complex) p S9 Pulse-gradient stimulated spin-echo NMR studies p S11 Table of Percentage of Hetero-complex Formation p S12 References p S13 S2 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 Experimental section General All reagents were purchased from Aldrich Chemical Company and used as received without further purification. Host 1 and 2 were synthesized as previously reported.1,2 NMR spectra were recorded on a Varian Inova 500 MHz spectrometer. Chemical shifts are reported relative to D2O (4.80 ppm). Endo Me c O O O O O O O O O O g O O g O O O d O d HO O HO O HO f O OH OH HO f O OH OH O b O O b O H H H H H H H H O e O e O O O O O O O O O O O O O O j j H H a H H H Ha H H m l m l HO O HO O O OH O OH HO O HO O O OH O OH 1 2 S3 Electronic Supplementary Material (ESI) for Chemical Communications This journal is © The Royal Society of Chemistry 2012 1H-NMR spectra of host 1, 2 and mixture of hosts in the absence of guests Figure S1: 1H NMR spectra of: 1) 1 mM of 1; 2) 1 mM of 2; 3) 0.5 mM of both 1 and 2 (all 10 mM sodium tetraborate solution). -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
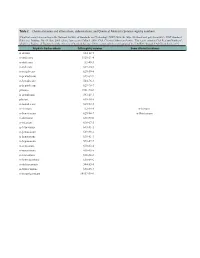
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No.