Genome-Wide Association Study Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Identification and Analysis of Prognostic Features in Human Cancers
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446243; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Genome-wide identification and analysis of prognostic features in human cancers Joan C. Smith1,2 and Jason M. Sheltzer1* 1. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724. 2. Google, Inc., New York, NY 10011. * Lead contact; to whom correspondence may be addressed. E-mail: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2021.06.01.446243; this version posted June 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Clinical decisions in cancer rely on precisely assessing patient risk. To improve our ability to accurately identify the most aggressive malignancies, we constructed genome-wide survival models using gene expression, copy number, methylation, and mutation data from 10,884 patients with known clinical outcomes. We identified more than 100,000 significant prognostic biomarkers and demonstrate that these genomic features can predict patient outcomes in clinically-ambiguous situations. While adverse biomarkers are commonly believed to represent cancer driver genes and promising therapeutic targets, we show that cancer features associated with shorter survival times are not enriched for either oncogenes or for successful drug targets. -

Characterization of a Monoclonal Antibody Panel Shows That the Myotonic Dystrophy Protein Kinase, DMPK, Is Expressed Almost Exclusively in Muscle and Heart
© 2000 Oxford University Press Human Molecular Genetics, 2000, Vol. 9, No. 14 2167–2173 Characterization of a monoclonal antibody panel shows that the myotonic dystrophy protein kinase, DMPK, is expressed almost exclusively in muscle and heart L.T. Lam, Y.C.N. Pham, Nguyen thi Man and G.E. Morris+ MRIC Biochemistry Group, PP18, North East Wales Institute, Mold Road, Wrexham LL11 2AW, UK Received 11 May 2000; Accepted 28 June 2000 DDBJ/EMBL/GenBank accession no. AF250871 Myotonic dystrophy (DM) is a multisystemic disorder ness), brain (mental retardation), eyes (cataracts), testis caused by an inherited CTG repeat expansion which (atrophy) and heart (conduction defects) (10). It is important, affects three genes encoding the DM protein kinase therefore, to establish the normal tissue distribution of all three (DMPK), a homeobox protein Six5 and a protein proteins produced from the DM locus. containing WD repeats. Using a panel of 16 mono- The human DMPK cDNA has 15 exons and predicts a ∼ clonal antibodies against several different DMPK protein of 70 kDa, although there may be variation in the size epitopes we detected DMPK, as a single protein of and sequence of the first exon (1–3,11,12). DMPK is a serine/ threonine protein kinase in which the catalytic domain ∼80 kDa, only in skeletal muscle, cardiac muscle and, (∼43 kDa) is followed by a coiled-coil domain (∼12 kDa) and to a lesser extent, smooth muscle. Many earlier a hydrophobic C-terminal domain. Alternative splicing has reports of DMPK with different sizes and tissue distri- been observed in a short VSGGG sequence between domains butions appear to be due to antibody cross-reactions and in the C-terminal domain (13). -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

In Silico Analysis of IDH3A Gene Revealed Novel Mutations Associated with Retinitis Pigmentosa
bioRxiv preprint doi: https://doi.org/10.1101/554196; this version posted February 18, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. In silico analysis of IDH3A gene revealed Novel mutations associated with Retinitis Pigmentosa Thwayba A. Mahmoud1*, Abdelrahman H. Abdelmoneim1, Naseem S. Murshed1, Zainab O. Mohammed2, Dina T. Ahmed1, Fatima A. Altyeb1, Nuha A. Mahmoud3, Mayada A. Mohammed1, Fatima A. Arayah1, Wafaa I. Mohammed1, Omnia S. Abayazed1, Amna S. Akasha1, Mujahed I. Mustafa1,4, Mohamed A. Hassan1 1- Department of Biotechnology, Africa city of Technology, Sudan 2- Hematology Department, Ribat University Hospital, Sudan 3- Biochemistry Department, faculty of Medicine, National University, Sudan 4- Department of Biochemistry, University of Bahri, Sudan *Corresponding Author: Thwayba A. Mahmoud, Email: [email protected] Abstract: Background: Retinitis Pigmentosa (RP) refers to a group of inherited disorders characterized by the death of photoreceptor cells leading to blindness. The aim of this study is to identify the pathogenic SNPs in the IDH3A gene and their effect on the structure and function of the protein. Method: we used different bioinformatics tools to predict the effect of each SNP on the structure and function of the protein. Result: 20 deleterious SNPs out of 178 were found to have a damaging effect on the protein structure and function. Conclusion: this is the first in silico analysis of IDH3A gene and 20 novel mutations were found using different bioinformatics tools, and they could be used as diagnostic markers for Retinitis Pigmentosa. -

A High Throughput, Functional Screen of Human Body Mass Index GWAS Loci Using Tissue-Specific Rnai Drosophila Melanogaster Crosses Thomas J
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. Baranski Washington University School of Medicine in St. Louis Aldi T. Kraja Washington University School of Medicine in St. Louis Jill L. Fink Washington University School of Medicine in St. Louis Mary Feitosa Washington University School of Medicine in St. Louis Petra A. Lenzini Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Baranski, Thomas J.; Kraja, Aldi T.; Fink, Jill L.; Feitosa, Mary; Lenzini, Petra A.; Borecki, Ingrid B.; Liu, Ching-Ti; Cupples, L. Adrienne; North, Kari E.; and Province, Michael A., ,"A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses." PLoS Genetics.14,4. e1007222. (2018). https://digitalcommons.wustl.edu/open_access_pubs/6820 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Thomas J. Baranski, Aldi T. Kraja, Jill L. Fink, Mary Feitosa, Petra A. Lenzini, Ingrid B. Borecki, Ching-Ti Liu, L. Adrienne Cupples, Kari E. North, and Michael A. Province This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6820 RESEARCH ARTICLE A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. -

Leveraging Tissue Specific Gene Expression Regulation to Identify Genes Associated with Complex Diseases
bioRxiv preprint doi: https://doi.org/10.1101/529297; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Leveraging tissue specific gene expression regulation to identify genes associated with complex diseases Wei Liu1,#, Mo Li2,#, Wenfeng Zhang2, Geyu Zhou1, Xing Wu4, Jiawei Wang1, Hongyu Zhao1,2,3* 1 Program of Computational Biology and Bioinformatics, Yale University, New Haven, CT, USA 06510 2 Department of Biostatistics, Yale School of Public Health, New Haven, CT, USA 06510 3 Department of Genetics, Yale School of Medicine, New Haven, CT, USA 06510 4 Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT, USA 06510 # These authors contributed equally to this work * To Whom correspondence should be addressed: Dr. Hongyu Zhao Department of Biostatistics Yale School of Public Health 60 College Street, New Haven, CT, 06520, USA [email protected] Key words: GWAS; gene expression imputation; Alzheimer’s disease; gene-level association test 1 bioRxiv preprint doi: https://doi.org/10.1101/529297; this version posted January 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract To increase statistical power to identify genes associated with complex traits, a number of methods like PrediXcan and FUSION have been developed using gene expression as a mediating trait linking genetic variations and diseases. -

Searching for Candidate Genes for Male Infertility
9/17/2015 :: Asian Journal of Andrology :: Home | Archive | AJA @ Nature | Online Submission | News & Events | Subscribe | APFA | Society | Links | Contact Us | 中文版 Pubmed Searching for candidate genes for male infertility B. N. Truong1, E.K. Moses2, J.E. Armes3, D.J. Venter3,4, H.W.G. Baker1 1Melbourne IVF Centre, 2Pregnancy Research Centre, Department of Obstetrics and Gynaecology, Royal Women's Hospital, University of Melbourne, Victoria 3053, Australia 3Departments of Pathology and Obstetrics/Gynaecology, University of Melbourne, Victoria 3010, Australia 4Murdoch Children's Research Institute, Royal Children's Hospital, Victoria 3052, Australia Asian J Androl 2003 Jun; 5: 137147 Keywords: male infertility; genetics; spermatogenesis; human genome; database; tissues; array Abstract Aim: We describe an approach to search for candidate genes for male infertility using the two human genome databases: the public University of California at Santa Cruz (UCSC) and private Celera databases which list known and predicted gene sequences and provide related information such as gene function, tissue expression, known mutations and single nucleotide polymorphisms (SNPs). Methods and Results: To demonstrate this in silico research, the following male infertility candidate genes were selected: (1) human BOULE, mutations of which may lead to germ cell arrest at the primary spermatocyte stage, (2) mutations of casein kinase 2 alpha genes which may cause globozoospermia, (3) DMRN9 which is possibly involved in the spermatogenic defect of myotonic dystrophy and (4) several testes expressed genes at or near the breakpoints of a balanced translocation associated with hypospermatogenesis. We indicate how information derived from the human genome databases can be used to confirm these candidate genes may be pathogenic by studying RNA expression in tissue arrays using in situ hybridization and gene sequencing. -

Exome Sequencing of 457 Autism Families Recruited Online Provides Evidence for Autism Risk Genes
www.nature.com/npjgenmed ARTICLE OPEN Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes Pamela Feliciano1, Xueya Zhou 2, Irina Astrovskaya 1, Tychele N. Turner 3, Tianyun Wang3, Leo Brueggeman4, Rebecca Barnard5, Alexander Hsieh 2, LeeAnne Green Snyder1, Donna M. Muzny6, Aniko Sabo6, The SPARK Consortium, Richard A. Gibbs6, Evan E. Eichler 3,7, Brian J. O’Roak 5, Jacob J. Michaelson 4, Natalia Volfovsky1, Yufeng Shen 2 and Wendy K. Chung1,8 Autism spectrum disorder (ASD) is a genetically heterogeneous condition, caused by a combination of rare de novo and inherited variants as well as common variants in at least several hundred genes. However, significantly larger sample sizes are needed to identify the complete set of genetic risk factors. We conducted a pilot study for SPARK (SPARKForAutism.org) of 457 families with ASD, all consented online. Whole exome sequencing (WES) and genotyping data were generated for each family using DNA from saliva. We identified variants in genes and loci that are clinically recognized causes or significant contributors to ASD in 10.4% of families without previous genetic findings. In addition, we identified variants that are possibly associated with ASD in an additional 3.4% of families. A meta-analysis using the TADA framework at a false discovery rate (FDR) of 0.1 provides statistical support for 26 ASD risk genes. While most of these genes are already known ASD risk genes, BRSK2 has the strongest statistical support and reaches genome-wide significance as a risk gene for ASD (p-value = 2.3e−06). -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Methylation of the G-C Rich Dmpk Gene: Causation For
METHYLATION OF THE G-C RICH DMPK GENE: CAUSATION FOR THE SEVERITY DIFFERENCE BETWEEN ADULT- ONSET AND CONGENITAL MYOTONIC DYSTROPHY? by Brian Barnette A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Honors Bachelor of Science in Biological Sciences. Spring 2010 Copyright 2010 Brian Barnette All Rights Reserved METHYLATION OF THE G-C RICH DMPK GENE: CAUSATION FOR THE SEVERITY DIFFERENCE BETWEEN ADULT- ONSET AND CONGENITAL MYOTONIC DYSTROPHY? by Brian Barnette Approved: __________________________________________________________ Vicky Funanage, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Grace Hobson, Ph.D. Committee member from the Department of Biological Sciences Approved: __________________________________________________________ Marlene Emara, Ph.D. Committee member from the Board of Senior Thesis Readers Approved: __________________________________________________________ Alan Fox, Ph.D. Director, University Honors Program ACKNOWLEDGMENTS This study could not have been possible without the help of several individuals. I would first like to thank Dr. Funanage for agreeing to sponsor not only my undergraduate research at the University of Delaware, but also for being my thesis advisor. Dr. Funanage had accepted me into her lab after a brief interview the winter of my sophomore year. Since then, I have greatly enjoyed my time in the Musculoskeletal Inherited Disease (MID) Laboratory, it was one of the greatest experiences I have had at the University of Delaware. I would also like to thank two individuals I met the same day as my interview with Dr. Funanage, Sarah Swain and Susan Kirwin. Sarah Swain was instrumental in not only in the writing of my first grant proposal, but also in my laboratory training. -

Alsaai Udel 0060D 14351.Pdf
INVESTIGATION OF TDRD7 FUNCTION IN OCULAR LENS DEVELOPMENT by Salma Mohammed Al Saai A dissertation submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biological Sciences Summer 2020 © 2020 Salma Mohammed Al Saai All Rights Reserved . INVESTIGATION OF TDRD7 FUNCTION IN OCULAR LENS DEVELOPMENT by Salma Mohammed Al Saai Approved: __________________________________________________________ Velia M. Fowler, Ph.D. Chair of the Department of Biological Sciences Approved: __________________________________________________________ John A. Pelesko, Ph.D. Dean of the College of Arts & Sciences Approved: __________________________________________________________ Douglas J. Doren, Ph.D. Interim Vice Provost for Graduate and Professional Education and Dean of the Graduate College I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Salil A. Lachke, Ph.D. Professor in charge of dissertation I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Melinda K. Duncan, Ph.D. Member of dissertation committee I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Donna Woulfe, Ph.D. Member of dissertation committee I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. -
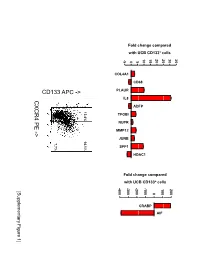
Supplementary Data
Fold change compared with UCB CD133+ cells 10 15 20 25 30 35 -5 0 5 COL4A1 CD68 PLAUR IL8 CXCR4 PE ADFP 13.4% 14.1% TFGBI PE NUPR - MMP12 > JUNB 7.7% SPP1 HDAC1 Fold change compared with UCB CD133+ cells -400 -300 -200 -100 100 200 [Supplementary Figure1] 0 CRABP AIF Rutella S et al. Supplementary Materials and Methods Real-time PCR Real-time quantitative PCR (qPCR) was performed on mRNA samples isolated from either endometrial cancer-derived CD133+ cells or umbilical cord blood CD133+ cells with the RNeasy plus Mini Kit (QIAGEN, Hilden, Germany), as already detailed. Quantitative PCR for a selected group of genes was performed on the iCycler iQ system (Bio-Rad, Hercules, CA). Primer sets were designed using the Beacon Design Software (Version 3) and the sequences available in the GeneBankTM data base. The specific oligonucleotide primer sequences are detailed in Supplementary Table 8. Complementary DNA (cDNA) was prepared starting from 1μg of total RNA using the iScrypt cDNA Synthesis Kit, which contains RNase H + MMLV reverse transcriptase random primers and 5x reaction mixture (Bio-Rad), according to the manufacturer’s instructions. Reactions were conducted in the PTC-0200 DNA Engine (MJ RESEARCH). Amplification was carried out in a total volume of 25μl containing 0.3μM of each specific primer, 12,5μl 2X SYBR Green Master Mix (100mM KCl, 40mM Tris-HCl, pH 8.4, 0.4mM of each dNTP, iTaq DNA polymerase, 6mM MgCl2, SYBR Green I, 20nM fluorescein, stabilizers; Bio-Rad) and 2μl of diluted cDNA. The PCR reactions were cycled starting with a 3-minute template denaturation step at 95°C followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C.