Arrhythmia Mutations in Calmodulin Cause Conformational Changes That Affect Interactions with the Cardiac Voltage-Gated Calcium Channel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Supplemental Material
Supplemental Table B ARGs in alphabetical order Symbol Title 3 months 6 months 9 months 12 months 23 months ANOVA Direction Category 38597 septin 2 1557 ± 44 1555 ± 44 1579 ± 56 1655 ± 26 1691 ± 31 0.05219 up Intermediate 0610031j06rik kidney predominant protein NCU-G1 491 ± 6 504 ± 14 503 ± 11 527 ± 13 534 ± 12 0.04747 up Early Adult 1G5 vesicle-associated calmodulin-binding protein 662 ± 23 675 ± 17 629 ± 16 617 ± 20 583 ± 26 0.03129 down Intermediate A2m alpha-2-macroglobulin 262 ± 7 272 ± 8 244 ± 6 290 ± 7 353 ± 16 0.00000 up Midlife Aadat aminoadipate aminotransferase (synonym Kat2) 180 ± 5 201 ± 12 223 ± 7 244 ± 14 275 ± 7 0.00000 up Early Adult Abca2 ATP-binding cassette, sub-family A (ABC1), member 2 958 ± 28 1052 ± 58 1086 ± 36 1071 ± 44 1141 ± 41 0.05371 up Early Adult Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A 136 ± 8 147 ± 6 147 ± 13 155 ± 9 185 ± 13 0.01272 up Midlife Acadl acetyl-Coenzyme A dehydrogenase, long-chain 423 ± 7 456 ± 11 478 ± 14 486 ± 13 512 ± 11 0.00003 up Early Adult Acadvl acyl-Coenzyme A dehydrogenase, very long chain 426 ± 14 414 ± 10 404 ± 13 411 ± 15 461 ± 10 0.01017 up Late Accn1 amiloride-sensitive cation channel 1, neuronal (degenerin) 242 ± 10 250 ± 9 237 ± 11 247 ± 14 212 ± 8 0.04972 down Late Actb actin, beta 12965 ± 310 13382 ± 170 13145 ± 273 13739 ± 303 14187 ± 269 0.01195 up Midlife Acvrinp1 activin receptor interacting protein 1 304 ± 18 285 ± 21 274 ± 13 297 ± 21 341 ± 14 0.03610 up Late Adk adenosine kinase 1828 ± 43 1920 ± 38 1922 ± 22 2048 ± 30 1949 ± 44 0.00797 up Early -
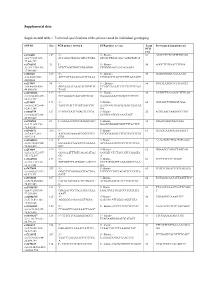
Supplemental Data Supplemental Table 1. Technical Specifications of the Primers Used for Individual Genotyping
Supplemental data Supplemental table 1. Technical specifications of the primers used for individual genotyping SNP ID Size PCR primer forward PCR primer reverse Temp Pyrosequencing primer(s) PCR (°C) rs776108 127 5’- 5’- Biotin- 61 ATACCTCTCATTTTGCAG chr3:77,825,927- ACCAGGCTAGGCATGCTATA GTCACTTAACAGCAGTGTGTCA 77,826,729 rs3746192 92 5’- 5’- Biotin- 56 AGGCTTGTAACCTGGA chr19:17,946,368- ATTCTAGGTGGCATGAGGG CTGGGGAGCAACAGAAGA 17,946,488 rs988147 169 5' - 5' - Biotine- 53 GCAGGGGGTAGAAATG chr6:45282108- AGCCATTAAAGAATTTCAAA TTGGATTTTATTCTTGTAATAGG 45282608 rs227849 94 5' - 5' - Biotine- 56 GGTTTAAGGTCTTTGCAT chr6:44,806,436- AGGAAAATAAACATGTGGTT TCTACCAATATTTTCTTTCGTAG 44,806,936 TAAG T rs10733833 127 5' - 5' - Biotine- 58 CATGTTTAAAACCTTTCAG chr10:68,418,227- GCCAAAACCAACAGTTCAT GAAAAAAATTGCACCTGTCTC 68,418,727 rs322609 197 5’- 5’-Biotin- 60 GGTAGCTGTGGGTGGA chr16:62,432,604- TAGTTGATTTTGCCAACCTG AAATGGGTGACAGAAGTAATAA 62,433,104 GA rs1884779 127 5’-TGGCTATTGGAGTTCTCA 5’-Biotin- 55 AGTGAATTAAGGGCTTGT chr20:45,857,969- CCATCCATCCCAAATAGT 45,858,469 rs4703908 83 5’- GAAAATGCCCAAGGTGAC 5’-Biotin- 52 GTGACAGTGGGCAAA chr5:71,802,353- GAATGTGGGTGTGTTTTACTCT 71,802,853 rs6946871 290 5’- 5’-Biotin- 61 GGAGGAAAGGAAAAGTT chr7:4,037,310- ATCAGATAAAATCGGCTTCT TCGGGAAGGTTTTTGTACTTTTG 4,037,810 GTG rs11865033 123 5’- 5’-Biotin- 61 AAAGTCTCTTCCTATGAGC chr16:78,082,945- AATAAACCAAGCCCTGAAAA ACTAAAATCCCCCTTTCCTCCA 78,083,445 GTC rs247004 170 5’- 5’-Biotin- 62 GGAAGCCAGACTAGCAG chr5:131,372,007- GGGGAATTTGTCAGAGATAG GGGATCCTCTACCATCCAAATA 131,372,507 GG -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Disease Mutations in the Ryanodine Receptor N-Terminal Region Couple to a Mobile Intersubunit Interface
ARTICLE Received 1 Oct 2012 | Accepted 15 Jan 2013 | Published 19 Feb 2013 DOI: 10.1038/ncomms2501 OPEN Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface Lynn Kimlicka1, Kelvin Lau1, Ching-Chieh Tung1 & Filip Van Petegem1 Ryanodine receptors are large channels that release Ca2 þ from the endoplasmic and sar- coplasmic reticulum. Hundreds of RyR mutations can cause cardiac and skeletal muscle disorders, yet detailed mechanisms explaining their effects have been lacking. Here we compare pseudo-atomic models and propose that channel opening coincides with widen- ing of a cytoplasmic vestibule formed by the N-terminal region, thus altering an interface targeted by 20 disease mutations. We solve crystal structures of several disease mutants that affect intrasubunit domain–domain interfaces. Mutations affecting intrasubunit ionic pairs alter relative domain orientations, and thus couple to surrounding interfaces. Buried disease mutations cause structural changes that also connect to the intersubunit contact area. These results suggest that the intersubunit contact region between N-terminal domains is a prime target for disease mutations, direct or indirect, and we present a model whereby ryanodine receptors and inositol-1,4,5-trisphosphate receptors are activated by altering domain arrangements in the N-terminal region. 1 Department of Biochemistry and Molecular Biology, Life Sciences Institute, University of British Columbia, Vancouver, British Columbia, Canada V6T 1Z3. Correspondence and requests for materials should be addressed to F.V.P. (email: fi[email protected]). NATURE COMMUNICATIONS | 4:1506 | DOI: 10.1038/ncomms2501 | www.nature.com/naturecommunications 1 & 2013 Macmillan Publishers Limited. All rights reserved. -

Physiological and Pathophysiological Regulation of the Ryanodine Receptor in Skeletal Muscle
Physiological and pathophysiological regulation of the ryanodine receptor in skeletal muscle Alisa Umanskaya Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015 © 2015 Alisa Umanskaya All rights reserved Abstract Physiological and pathophysiological regulation of ryanodine receptor in skeletal muscle Alisa Umanskaya Ryanodine receptor calcium release channels are essential for skeletal muscle contraction, as they mediate the release of calcium ions from intracellular stores into the cytosol. The data presented in this dissertation demonstrate the evolutionarily conserved mechanisms of skeletal muscle ryanodine receptor regulation in the physiological and pathophysiological states. Adrenergic stimulation causes increased skeletal muscle force, however, despite the well- established role of this physiological response, the molecular mechanism is not known. Here we present a mechanism whereby phosphorylation of a single amino acid on the ryanodine receptor is a key signal in the physiological stress-induced inotropic response in mouse skeletal muscle. Therefore acute post-translational modifications of ryanodine receptor channels are important for healthy muscle contraction. Conversely, chronic stress-induced post-translational modifications result in poorly functioning murine ryanodine receptor channels that contribute to skeletal muscle dysfunction in age- dependent skeletal muscle weakness and Muscular Dystrophies. Finally, we present data that demonstrates striking evolutionary conservation in ryanodine receptor regulation in the physiological and pathophysiological states between mice and C. elegans. This work has broad implications for understanding the underlying mechanisms of skeletal muscle contraction and important disorders that affect human health. Furthermore, this works presents ryanodine receptor channels as a viable therapeutic target for age-related skeletal muscle weakness, Muscular Dystrophies, and also implicates C. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

RYR2 Gene Ryanodine Receptor 2
RYR2 gene ryanodine receptor 2 Normal Function The RYR2 gene provides instructions for making a protein called ryanodine receptor 2. This protein is part of a family of ryanodine receptors, which form channels that transport positively charged calcium atoms (calcium ions) within cells. Channels made with the ryanodine receptor 2 protein are found in heart (cardiac) muscle cells called myocytes. These channels are embedded in the outer membrane of a cell structure called the sarcoplasmic reticulum, which acts as a storage center for calcium ions. The RYR2 channel controls the flow of calcium ions out of the sarcoplasmic reticulum. For the heart to beat normally, the cardiac muscle must tense (contract) and relax in a coordinated way. This cycle of muscle contraction and relaxation results from the precise control of calcium ions within myocytes. In response to certain signals, the RYR2 channel releases calcium ions from the sarcoplasmic reticulum into the surrounding cell fluid (the cytoplasm). The resulting increase in calcium ion concentration triggers the cardiac muscle to contract, which pumps blood out of the heart. Calcium ions are then transported back into the sarcoplasmic reticulum, and the cardiac muscle relaxes. In this way, the release and reuptake of calcium ions in myocytes produces a regular heart rhythm. Health Conditions Related to Genetic Changes Catecholaminergic polymorphic ventricular tachycardia More than 200 mutations in the RYR2 gene have been found to cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a heart condition characterized by an abnormal heart rhythm (arrhythmia) during exercise or emotional stress, which can be fatal. Almost all of the RYR2 gene mutations involved in CPVT change single protein building blocks (amino acids) in the ryanodine receptor 2 protein. -

MALAT1 Knockdown Protects from Bronchial/Tracheal Smooth Muscle Cell Injury Via Regulation of Microrna-133A/Ryanodine Receptor 2 Axis
J Biosci (2021)46:28 Ó Indian Academy of Sciences DOI: 10.1007/s12038-021-00149-3 (0123456789().,-volV)(0123456789().,-volV) MALAT1 knockdown protects from bronchial/tracheal smooth muscle cell injury via regulation of microRNA-133a/ryanodine receptor 2 axis 1 2 MINGZHU YANG and LI WANG * 1Department of Pediatric Internal Medicine, Xian Yang Central Hospital, Xianyang 712000, People’s Republic of China 2Department of Laboratory Medicine, Affiliated Hospital of Shaanxi University of Chinese Medicine, Xianyang 712000, People’s Republic of China *Corresponding author (Email, [email protected]) MS received 9 December 2019; accepted 12 February 2021 Asthma has significant impacts on living quality particularly in children. Long noncoding RNA (lncRNA) MALAT1 plays a crucial role in neonatal respiratory diseases. Meanwhile, MALAT1 knockdown could induce viability and attenuate apoptosis of airway-related cells. However, the role of MALAT1 in neonatal asthma, asthma-related cell, and its possible mechanism is unclear. This study aims to investigate MALAT1 level in asthma and to identify the effects of MALAT1 on bronchial/tracheal smooth muscle cells (B/TSMCs). Newborn asthma modeling rat was constructed by introducing ovalbumin (OVA). MALAT1 levels in tissues or B/TSMCs were determined by RT-qPCR. Exogenous changes of MALAT1, RyR2 or miR-133a in B/TSMCs were fulfilled by cell transfection; cell apoptosis was measured by using Cell Death Detection ELISA kit and Hochest33342; IL-6, TNF-a and IL-1b level was detected by using corresponding ELISA kit; ryanodine receptor 2 (RyR2) mRNA and miR-133a level was determined by RT-qPCR; cleaved caspase-3 (c-caspase-3) and RyR2 expression was detected by Western blot; luciferase reporter assay was performed to confirm the target regulation of miR-133a on RyR2. -
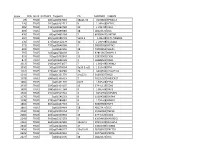
Supplementary Table 2
Index CPG_ISLANDDISTANCE_TO_TSSTargetID Chr MAPINFO SYMBOL 29 TRUE 185 cg00027083 18p11.32 5533801 EPB41L3 220 TRUE 141 cg00202711 X 1.14E+08 HTR2C 285 TRUE 446 cg00263760 10 1.19E+08 VAX1 403 TRUE cg00399483 18 48122376 DCC 476 TRUE 43 cg00465284 2 80385409 LRRTM1 504 TRUE 691 cg00489401 5q35.3 1.8E+08 FLT4/VEGFR3 530 TRUE 275 cg00512279 10 1.19E+08 SLC18A2 571 TRUE 779 cg00548268 7 98083766 NPTX2 692 TRUE cg00662556 18 73092352 GALR1 797 TRUE 103 cg00768439 X 92815367 NAP1L3 815 TRUE 79 cg00792849 15 32833902 CX36 874 TRUE 193 cg00848728 1 58488606 DAB1 1013 TRUE 196 cg00973677 7 1.36E+08 CHRM2 1036 TRUE 50 cg01009664 3q13.3-q21 1.31E+08 TRH 1161 TRUE 325 cg01144286 20 9443596 C20orf103 1241 TRUE 59 cg01231779 14q22.1 51605637 NID2 1378 TRUE 698 cg01366419 7 70235027 WBSCR17 1404 TRUE cg01401376 6q23 1.34E+08 EYA4 1435 TRUE 204 cg01424107 13 27441113 CDX2 1493 TRUE 188 cg01471384 4 1.08E+08 DKK2 1542 TRUE 191 cg01519742 4 6252992 JAKMIP1 1570 TRUE cg01546563 8 11604598 GATA4 1598 TRUE 578 cg01580681 4 1.75E+08 HAND2 1863 TRUE 684 cg01837719 6 96569906 FUT9 1865 TRUE cg01839464 18 48122475 DCC 2040 TRUE 295 cg02008154 7 35260062 TBX20 2082 TRUE 203 cg02055963 13 27441520 CDX2 2243 TRUE 565 cg02217159 6 63054656 KHDRBS2 2261 TRUE 363 cg02233559 13q12.3 28191325 SLC46A3 2442 TRUE 47 cg02422694 17 44010789 HOXB4 2456 TRUE 89 cg02440177 19q13.41 58188507 ZNF702 2625 TRUE 98 cg02622316 6 28475389 ZNF96 2627 TRUE cg02624705 18 48122676 DCC 2776 TRUE 657 cg02774439 4 1.75E+08 HAND2 2848 TRUE 41 cg02847500 X 1.39E+08 SOX3 2924 TRUE 49 cg02919422 -

Original Article Unbalanced Upregulation of Ryanodine Receptor 2 Plays a Particular Role in Early Development of Daunorubicin Cardiomyopathy
Am J Transl Res 2015;7(7):1280-1294 www.ajtr.org /ISSN:1943-8141/AJTR0010909 Original Article Unbalanced upregulation of ryanodine receptor 2 plays a particular role in early development of daunorubicin cardiomyopathy Dana Kucerova1,2, Gabriel Doka1, Peter Kruzliak3, Katarina Turcekova1, Jana Kmecova1, Zuzana Brnoliakova4, Jan Kyselovic1, Uwe Kirchhefer2, Frank U Müller2, Peter Krenek1, Peter Boknik2, Jan Klimas1 1Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University, Bratislava, Slovak Re- public; 2Institut für Pharmakologie und Toxikologie, Universitätsklinikum Münster, Münster, Germany; 3Internation- al Clinical Research Center, St. Anne’s University Hospital and Masaryk University, Brno, Czech Republic; 4Institute of Experimental Pharmacology, Slovak Academy of Sciences, Bratislava, Slovak Republic Received May 31, 2015; Accepted July 14, 2015; Epub July 15, 2015; Published July 30, 2015 Abstract: Calcium release channel on the sarcoplasmic reticulum of cardiomyocytes (ryanodine receptor type 2, RyR2) plays a critical role in the regulation of calcium and was identified as a crucial factor for development of chronic anthracycline cardiomyopathy. Its early stages are less well described although these determine the later development. Hence, we tested the effect of repeated, short-term anthracycline (daunorubicin) administration on cardiac performance, cardiomyocyte function and accompanied changes in calcium regulating proteins expression. Ten-twelve weeks old male Wistar rats were administered with 6 doses of daunorubicin (DAU, 3 mg/kg, i.p., every 48 h), controls (CON) received vehicle. Left ventricular function (left ventricular pressure, LVP; rate of pressure de- velopment, +dP/dt and decline, -dP/dt) was measured using left ventricular catheterization under tribromethanol anaesthesia (15 ml/kg b.w.).