Supplemental Materials Supplemental Methods Generation of Dsg1a And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ONCOGENOMICS Through the Use of Chemotherapeutic Agents
Oncogene (2002) 21, 7749 – 7763 ª 2002 Nature Publishing Group All rights reserved 0950 – 9232/02 $25.00 www.nature.com/onc Expression profiling of primary non-small cell lung cancer for target identification Jim Heighway*,1, Teresa Knapp1, Lenetta Boyce1, Shelley Brennand1, John K Field1, Daniel C Betticher2, Daniel Ratschiller2, Mathias Gugger2, Michael Donovan3, Amy Lasek3 and Paula Rickert*,3 1Target Identification Group, Roy Castle International Centre for Lung Cancer Research, University of Liverpool, 200 London Road, Liverpool L3 9TA, UK; 2Institute of Medical Oncology, University of Bern, 3010 Bern, Switzerland; 3Incyte Genomics Inc., 3160 Porter Drive, Palo Alto, California, CA 94304, USA Using a panel of cDNA microarrays comprising 47 650 Introduction transcript elements, we have carried out a dual-channel analysis of gene expression in 39 resected primary The lack of generally effective therapies for lung cancer human non-small cell lung tumours versus normal lung leads to the high mortality rate associated with this tissue. Whilst *11 000 elements were scored as common disease. Recorded 5-year survival figures vary, differentially expressed at least twofold in at least one to some extent perhaps reflecting differences in sample, 96 transcripts were scored as over-represented ascertainment methods, but across Europe are approxi- fourfold or more in at least seven out of 39 tumours mately 10% (Bray et al., 2002). Whilst surgery is an and 30 sequences 16-fold in at least two out of 39 effective strategy for the treatment of localized lesions, tumours, including 24 transcripts in common. Tran- most patients present with overtly or covertly dissemi- scripts (178) were found under-represented fourfold in nated disease. -

Downloaded the Normalized Gene Expression Profile from the GEO (Package Geoquery (5))
1 SUPPLEMENTARY MATERIALS 2 Supplementary Methods 3 Study design 4 The workflow to identify and validate the TME risk score and TME subtypes in gastric cancer is 5 depicted in Fig. S1. We first estimated the absolute abundance levels of the major stromal and 6 immune cell types in the TME using bulk gene expression data, and assessed the prognostic 7 effect of these cells in a discovery cohort. Next, we constructed a TME risk score and validated it 8 in two independent gene expression validation cohorts and three immunohistochemistry 9 validation cohorts. Finally, we stratified patients into four TME subtypes and examined their 10 genomic and molecular features and relation to established molecular subtypes. 11 Gene expression data 12 To explore the prognostic landscape of the TME, we used four gene expression profile (GEP) 13 datasets of resected gastric cancer patients with publicly available clinical information, namely, 14 ACRG (GSE62254) (1), GSE15459 (2), GSE84437 (3), and TCGA stomach adenocarcinoma 15 (STAD). Specifically, the raw microarray data in the ACRG cohort and GSE15459 were retrieved 16 from the Gene Expression Omnibus (GEO), and normalized by the RMA algorithm (package affy) 17 using custom chip definition files (Brainarray version 23 (4)) that convert Affymetrix probesets to 18 Entrez gene IDs. For GSE84437 dataset, which was measured by the Illumina platform, we 19 downloaded the normalized gene expression profile from the GEO (package GEOquery (5)). The 20 Illumina probes were also mapped to Entrez genes. For multiple probes mapping to the same 21 Entrez gene, we selected the one with the maximum mean expression level as the surrogate for 22 the Entrez gene using the function of collapseRows (6) (package WGCNA). -

Salivary Exrna Biomarkers to Detect Gingivitis and Monitor Disease Regression
Received: 3 December 2017 | Revised: 14 April 2018 | Accepted: 13 May 2018 DOI: 10.1111/jcpe.12930 OTHER Salivary exRNA biomarkers to detect gingivitis and monitor disease regression Karolina E. Kaczor-Urbanowicz1,2* | Harsh M. Trivedi3* | Patricia O. Lima1,4 | Paulo M. Camargo5 | William V. Giannobile6 | Tristan R. Grogan7 | Frederico O. Gleber-Netto8 | Yair Whiteman9 | Feng Li1 | Hyo Jung Lee10 | Karan Dharia11 | Katri Aro1 | Carmen Martin Carerras-Presas12 | Saarah Amuthan11 | Manjiri Vartak11 | David Akin1 | Hiba Al-adbullah11 | Kanika Bembey11 | Perry R. Klokkevold5 | David Elashoff7 | Virginia M. Barnes13 | Rose Richter13 | William DeVizio13 | James G. Masters3 | David T. W. Wong1 1UCLA School of Dentistry, Center for Oral/Head & Neck Oncology Research, University of California at Los Angeles, Los Angeles, California 2Section of Orthodontics, UCLA School of Dentistry, University of California at Los Angeles, Los Angeles, California 3Early Research Oral Care, Colgate Palmolive Co., Piscataway, New Jersey 4Department of Physiological Sciences, Piracicaba Dental School, University of Campinas, Piracicaba, São Paulo, Brazil 5Section of Periodontics, UCLA School of Dentistry, University of California at Los Angeles, Los Angeles, California 6Department of Periodontics and Oral Medicine, School of Dentistry, University of Michigan, Ann Arbor, Michigan 7Department of Biostatistics, University of California at Los Angeles, Los Angeles, California 8Medical Genomics Laboratory, Centro Internacional de Pesquisa e Ensino (CIPE), AC Camargo Cancer -

Arg206cys Substitution in DNASE1L3 Causes a Defect in DNASE1L3 Protein Secretion That Confers Risk of Systemic Lupus Erythematos
Ann Rheum Dis: first published as 10.1136/annrheumdis-2020-218810 on 17 January 2021. Downloaded from Systemic lupus erythematosus TRANSLATIONAL SCIENCE Arg206Cys substitution in DNASE1L3 causes a defect in DNASE1L3 protein secretion that confers risk of systemic lupus erythematosus Latanya N Coke ,1 Hongxiu Wen ,1 Mary Comeau ,2 Mustafa H Ghanem ,1,3 Andrew Shih ,1 Christine N Metz ,1,3 Wentian Li ,1 Carl D Langefeld ,2 Peter K Gregersen ,1,3 Kim R Simpfendorfer 1,3 Handling editor Josef S ABSTRACT Key messages Smolen Objectives To determine if the polymorphism encoding the Arg206Cys substitution in DNASE1L3 explains the ► Additional material is What is already known about this subject? published online only. To view, association of the DNASE1L3/PXK gene locus with systemic ► The coding polymorphism rs35677470 is the please visit the journal online lupus erythematosus (SLE) and to examine the effect of the causal variant for rheumatoid arthritis (RA) and (http:// dx. doi. org/ 10. 1136/ Arg206Cys sequence change on DNASE1L3 protein function. scleroderma/systemic sclerosis (SSc) association annrheumdis- 2020- 218810). Methods Conditional analysis for rs35677470 was in the DNASE1L3/PXK locus and is reported to 1 performed on cases and controls with European ancestry The Institute of Molecular eliminate DNASE1L3 enzyme function. Medicine, Northwell Health from the SLE Immunochip study, and genotype and haplotype Feinstein Institutes for Medical frequencies were compared. DNASE1L3 protein levels were What does this study add? Research, Manhasset, New measured in cells and supernatants of HEK293 cells and York, USA ► The coding polymorphism rs35677470 is 2 monocyte-derived dendritic cells expressing recombinant and Department of Biostatistics the causal variant for the systemic lupus endogenous 206Arg and 206Cys protein variants. -

De Novo Characterization of Cell-Free DNA Fragmentation Hotspots Boosts
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.16.201350; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 De novo characterization of cell-free DNA fragmentation hotspots 2 boosts the power for early detection and localization of multi- 3 cancer 4 Xionghui Zhou1, Yaping Liu1-4 * 5 1 Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 6 45229 7 2 Division of Biomedical Informatics, Cincinnati Children’s Hospital Medical Center, 8 Cincinnati, OH 45229 9 3 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH 10 45229 11 4 Department of Electrical Engineering and Computing Sciences, University of Cincinnati 12 College of Engineering and Applied Science, Cincinnati, OH 45229 13 * Email: [email protected] 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.16.201350; this version posted July 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 26 Abstract 27 The global variation of cell-free DNA fragmentation patterns is a promising biomarker for 28 cancer diagnosis. However, the characterization of its hotspots and aberrations in early- 29 stage cancer at the fine-scale is still poorly understood. -

Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity
Article Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity Graphical Abstract Authors Vanja Sisirak, Benjamin Sally, Vivette D’Agati, ..., Robert M. Clancy, Jill P. Buyon, Boris Reizis Correspondence [email protected] In Brief Extracellular microparticle-associated chromatin is a potential self-antigen normally digested by circulating DNASE1L3. The loss of this tolerance mechanism in mice and humans contributes to lupus, and the restoration of this mechanism may represent a therapeutic opportunity in the disease. Highlights d Rapid anti-DNA antibody response, followed by SLE in Dnase1l3-deficient mice d Autoreactivity is repressed by circulating DNASE1L3 and is independent of STING d DNASE1L3 digests genomic DNA in microparticles released from apoptotic cells d DNASE1L3 prevents autoantibody binding to chromatin on microparticle surface Sisirak et al., 2016, Cell 166, 88–101 June 30, 2016 ª 2016 Elsevier Inc. http://dx.doi.org/10.1016/j.cell.2016.05.034 Article Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity Vanja Sisirak,1,12 Benjamin Sally,1,2,12 Vivette D’Agati,3 Wilnelly Martinez-Ortiz,4 Z. Birsin O¨ zc¸ akar,5 Joseph David,1 Ali Rashidfarrokhi,1 Ada Yeste,6 Casandra Panea,2 Asiya Seema Chida,7 Milena Bogunovic,8 Ivaylo I. Ivanov,2 Francisco J. Quintana,6 Inaki Sanz,7 Keith B. Elkon,9 Mustafa Tekin,10 Fatosx Yalc¸ ınkaya,5 Timothy J. Cardozo,4 Robert M. Clancy,11 Jill P. Buyon,11 and Boris Reizis1,2,11,* 1Department of Pathology, New York University School of Medicine, New -

Digestion of Chromatin in Apoptotic Cell
Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity Vanja Sisirak, Benjamin Sally, Vivette d’Agati, Wilnelly Martinez-Ortiz, Z Birsin Özçakar, Joseph David, Ali Rashidfarrokhi, Ada Yeste, Casandra Panea, Asiya Chida, et al. To cite this version: Vanja Sisirak, Benjamin Sally, Vivette d’Agati, Wilnelly Martinez-Ortiz, Z Birsin Özçakar, et al.. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell, Elsevier, 2016, 166 (1), pp.88 - 101. 10.1016/j.cell.2016.05.034. hal-03045372 HAL Id: hal-03045372 https://hal.archives-ouvertes.fr/hal-03045372 Submitted on 27 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity Vanja Sisirak1*, Benjamin Sally1,2*, Vivette D’Agati3, Wilnelly Martinez-Ortiz4, Z. Birsin Özçakar5, Joseph David1, Ali Rashidfarrokhi1, Ada Yeste6, Casandra Panea2, Asiya Seema Chida8, Milena Bogunovic7, Ivaylo I. Ivanov2, Francisco J. Quintana6, Inaki Sanz8, Keith B. Elkon9, Mustafa Tekin10, Fatoş Yalçınkaya5, Timothy J. Cardozo4, Robert M. Clancy11, Jill P. Buyon11 and Boris Reizis1,2,11 1Dept. of Pathology, New York University School of Medicine, New York, NY; 2Dept. of Microbiology and Immunology, Columbia University Medical Center, New York, NY; 3Dept. -

Mouse Steap4 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Steap4 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Steap4 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Steap4 gene (NCBI Reference Sequence: NM_054098 ; Ensembl: ENSMUSG00000012428 ) is located on Mouse chromosome 5. 5 exons are identified, with the ATG start codon in exon 2 and the TAA stop codon in exon 5 (Transcript: ENSMUST00000115421). Exon 2~3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Steap4 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-212F6 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit adipose accumulation, oxidative stress, increased liver weight, lower metabolic rate, hypoactivity, insulin resistance, glucose intolerance, mild hyperglycemia and dyslipidemia. Exon 2 starts from about 100% of the coding region. The knockout of Exon 2~3 will result in frameshift of the gene. The size of intron 1 for 5'-loxP site insertion: 14897 bp, and the size of intron 3 for 3'-loxP site insertion: 1385 bp. The size of effective cKO region: ~2082 bp. The cKO region does not have any other known gene. Page 1 of 7 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Steap4 Homology arm cKO region loxP site Page 2 of 7 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Single-Cell Analysis Uncovers Fibroblast Heterogeneity
ARTICLE https://doi.org/10.1038/s41467-020-17740-1 OPEN Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination ✉ Lars Muhl 1,2 , Guillem Genové 1,2, Stefanos Leptidis 1,2, Jianping Liu 1,2, Liqun He3,4, Giuseppe Mocci1,2, Ying Sun4, Sonja Gustafsson1,2, Byambajav Buyandelger1,2, Indira V. Chivukula1,2, Åsa Segerstolpe1,2,5, Elisabeth Raschperger1,2, Emil M. Hansson1,2, Johan L. M. Björkegren 1,2,6, Xiao-Rong Peng7, ✉ Michael Vanlandewijck1,2,4, Urban Lendahl1,8 & Christer Betsholtz 1,2,4 1234567890():,; Many important cell types in adult vertebrates have a mesenchymal origin, including fibro- blasts and vascular mural cells. Although their biological importance is undisputed, the level of mesenchymal cell heterogeneity within and between organs, while appreciated, has not been analyzed in detail. Here, we compare single-cell transcriptional profiles of fibroblasts and vascular mural cells across four murine muscular organs: heart, skeletal muscle, intestine and bladder. We reveal gene expression signatures that demarcate fibroblasts from mural cells and provide molecular signatures for cell subtype identification. We observe striking inter- and intra-organ heterogeneity amongst the fibroblasts, primarily reflecting differences in the expression of extracellular matrix components. Fibroblast subtypes localize to discrete anatomical positions offering novel predictions about physiological function(s) and regulatory signaling circuits. Our data shed new light on the diversity of poorly defined classes of cells and provide a foundation for improved understanding of their roles in physiological and pathological processes. 1 Karolinska Institutet/AstraZeneca Integrated Cardio Metabolic Centre, Blickagången 6, SE-14157 Huddinge, Sweden. -
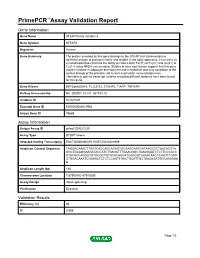
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name STEAP family member 4 Gene Symbol STEAP4 Organism Human Gene Summary The protein encoded by this gene belongs to the STEAP (six transmembrane epithelial antigen of prostate) family and resides in the golgi apparatus. It functions as a metalloreductase that has the ability to reduce both Fe(3+) to Fe(2+) and Cu(2+) to Cu(1+) using NAD(+) as acceptor. Studies in mice and human suggest that this gene maybe involved in adipocyte development and metabolism and may contribute to the normal biology of the prostate cell as well as prostate cancer progression. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp666D049, FLJ23153, STAMP2, TIARP, TNFAIP9 RefSeq Accession No. NC_000007.13, NT_007933.15 UniGene ID Hs.521008 Ensembl Gene ID ENSG00000127954 Entrez Gene ID 79689 Assay Information Unique Assay ID qHsaCID0021230 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000380079, ENST00000301959 Amplicon Context Sequence TAGGACAAACTTGATCACCAGCACAGTGCAAGGAATGATAAGCCCTAACACGTA GGCTGCAGGAAGATACCATCTGAGATTTGAAGGGCTGAGGAATCTCTTCCCACC GTACACCAGGGTGTGGGCTGTACACAAGATCAGGGTCAAATAACCCAGTTTGGA CTGGACAAATCGGAACTCTCTCCAGTTGACTGCATTGCTAACAGATGGCAAAGAA G Amplicon Length (bp) 188 Chromosome Location 7:87908782-87910285 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.999 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 22.3 cDNA Tm (Celsius) 84 gDNA Cq 38.74 Specificity (%) 100 Information