The Long Noncoding RNA HOTAIRM1 Controlled by AML1 Enhances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Molecular and Genetic Analysis of Otosclerosis
A molecular and genetic analysis of otosclerosis Joanna Lauren Ziff Submitted for the degree of PhD University College London January 2014 1 Declaration I, Joanna Ziff, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Where work has been conducted by other members of our laboratory, this has been indicated by an appropriate reference. 2 Abstract Otosclerosis is a common form of conductive hearing loss. It is characterised by abnormal bone remodelling within the otic capsule, leading to formation of sclerotic lesions of the temporal bone. Encroachment of these lesions on to the footplate of the stapes in the middle ear leads to stapes fixation and subsequent conductive hearing loss. The hereditary nature of otosclerosis has long been recognised due to its recurrence within families, but its genetic aetiology is yet to be characterised. Although many familial linkage studies and candidate gene association studies to investigate the genetic nature of otosclerosis have been performed in recent years, progress in identifying disease causing genes has been slow. This is largely due to the highly heterogeneous nature of this condition. The research presented in this thesis examines the molecular and genetic basis of otosclerosis using two next generation sequencing technologies; RNA-sequencing and Whole Exome Sequencing. RNA–sequencing has provided human stapes transcriptomes for healthy and diseased stapes, and in combination with pathway analysis has helped identify genes and molecular processes dysregulated in otosclerotic tissue. Whole Exome Sequencing has been employed to investigate rare variants that segregate with otosclerosis in affected families, and has been followed by a variant filtering strategy, which has prioritised genes found to be dysregulated during RNA-sequencing. -

Characterizing G-Quadruplex Mediated Regulation of the Amyloid Precursor Protein Expression by Ezekiel Matthew David Crenshaw Oc
I Characterizing G-Quadruplex Mediated Regulation of the Amyloid Precursor Protein Expression By Ezekiel Matthew David Crenshaw October, 2016 A Dissertation Presented to the Faculty of Drexel University Department of Biology In partial fulfillment of the Requirements for the Degree of Ph.D. Daniel Marenda, Ph.D. (Chair) Elias Spiliotis, Ph.D. Associate Professor, Associate Professor, Director, Graduate Student Program Director, Cell Imaging Center Co-Director, Cell Imaging Center Dept. Biology Dept. Biology Drexel University Drexel University Tali Gidalevitz, Ph.D. Aleister Saunders, Ph.D. (Advisor) Assistant Professor, Dept. Biology Associate Professor, Drexel University Director of the RNAi Resource Center, Dept. Biology Jeff Twiss, Ph.D. Senior Vice Provost for Research, Drexel University Professor, SmartState Chair, Childhood Neurotherapeutics Michael Akins, Ph.D.(Co-Advisor) Dept. Biology University of South Carolina Assistant Professor, Dept. Biology Drexel University II Acknowledgements Proverbs 3:5-6, “Trust in the Lord with all thine heart and lean not unto thine own understanding. In all thine ways acknowledge Him and He shall direct thy paths” I want to start off my acknowledging my LORD, who is known by many names. He is El Elyon, The Most High God; He is YAHWEH Shammah, for He has been there for me; He is YAHWEH Yireh, for He has provided for all of my needs; He is YAHWEH Shalom, for He is my peace. He is Yeshua Ha Mashiach, for He is my Savior. Coming from humble beginnings, my faith in the LORD has been the source of strength and encouragement to overcome the many obstacles I faced growing up, as well as in my pursuit for my Ph.D. -

Nucleolin and Its Role in Ribosomal Biogenesis
NUCLEOLIN: A NUCLEOLAR RNA-BINDING PROTEIN INVOLVED IN RIBOSOME BIOGENESIS Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Julia Fremerey aus Hamburg Düsseldorf, April 2016 2 Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Referent: Prof. Dr. A. Borkhardt Korreferent: Prof. Dr. H. Schwender Tag der mündlichen Prüfung: 20.07.2016 3 Die vorgelegte Arbeit wurde von Juli 2012 bis März 2016 in der Klinik für Kinder- Onkologie, -Hämatologie und Klinische Immunologie des Universitätsklinikums Düsseldorf unter Anleitung von Prof. Dr. A. Borkhardt und in Kooperation mit dem ‚Laboratory of RNA Molecular Biology‘ an der Rockefeller Universität unter Anleitung von Prof. Dr. T. Tuschl angefertigt. 4 Dedicated to my family TABLE OF CONTENTS 5 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................... 5 LIST OF FIGURES ......................................................................................................10 LIST OF TABLES .......................................................................................................12 ABBREVIATION .........................................................................................................13 ABSTRACT ................................................................................................................19 ZUSAMMENFASSUNG -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

The Landscape of Human Mutually Exclusive Splicing
bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The landscape of human mutually exclusive splicing Klas Hatje1,2,#,*, Ramon O. Vidal2,*, Raza-Ur Rahman2, Dominic Simm1,3, Björn Hammesfahr1,$, Orr Shomroni2, Stefan Bonn2§ & Martin Kollmar1§ 1 Group of Systems Biology of Motor Proteins, Department of NMR-based Structural Biology, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany 2 Group of Computational Systems Biology, German Center for Neurodegenerative Diseases, Göttingen, Germany 3 Theoretical Computer Science and Algorithmic Methods, Institute of Computer Science, Georg-August-University Göttingen, Germany § Corresponding authors # Current address: Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland $ Current address: Research and Development - Data Management (RD-DM), KWS SAAT SE, Einbeck, Germany * These authors contributed equally E-mail addresses: KH: [email protected], RV: [email protected], RR: [email protected], DS: [email protected], BH: [email protected], OS: [email protected], SB: [email protected], MK: [email protected] - 1 - bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Identification of Genomic Targets of Krüppel-Like Factor 9 in Mouse Hippocampal
Identification of Genomic Targets of Krüppel-like Factor 9 in Mouse Hippocampal Neurons: Evidence for a role in modulating peripheral circadian clocks by Joseph R. Knoedler A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Neuroscience) in the University of Michigan 2016 Doctoral Committee: Professor Robert J. Denver, Chair Professor Daniel Goldman Professor Diane Robins Professor Audrey Seasholtz Associate Professor Bing Ye ©Joseph R. Knoedler All Rights Reserved 2016 To my parents, who never once questioned my decision to become the other kind of doctor, And to Lucy, who has pushed me to be a better person from day one. ii Acknowledgements I have a huge number of people to thank for having made it to this point, so in no particular order: -I would like to thank my adviser, Dr. Robert J. Denver, for his guidance, encouragement, and patience over the last seven years; his mentorship has been indispensable for my growth as a scientist -I would also like to thank my committee members, Drs. Audrey Seasholtz, Dan Goldman, Diane Robins and Bing Ye, for their constructive feedback and their willingness to meet in a frequently cold, windowless room across campus from where they work -I am hugely indebted to Pia Bagamasbad and Yasuhiro Kyono for teaching me almost everything I know about molecular biology and bioinformatics, and to Arasakumar Subramani for his tireless work during the home stretch to my dissertation -I am grateful for the Neuroscience Program leadership and staff, in particular -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation
Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Colognori, David A. 2019. Dissection of Xist Functional Elements Involved in X-Chromosome Inactivation. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42029476 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Dissection of Xist functional elements involved in X-chromosome inactivation A dissertation presented by David A. Colognori to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biological and Biomedical Sciences Harvard University Cambridge, Massachusetts May 2019 © 2019 by David A. Colognori All rights reserved. Dissertation Advisor: Jeannie T. Lee David A. Colognori Dissection of Xist functional elements involved in X-chromosome inactivation Abstract X-chromosome inactivation (XCI) is the epigenetic process of silencing one of the two X chromosomes in female mammals to balance X-linked gene dosage with that of males. This process is governed by the long noncoding RNA Xist. Xist is expressed from and coats one X chromosome in cis, leading to recruitment of various protein factors to silence gene expression. However, the functional RNA elements and molecular mechanisms involved in Xist coating and silencing remain ambiguous. -

Supplementary Table S4
Supplementary Table S4 - Genes downregulated by anti-MYCN PNA ProbeID Symbol Description RH30ctrl_SignalRH30mut__SignalRH30pna_Signalpna vs mut pna vs ctrl 210842_at NRP2 neuropilin 2 109.8 83.2 3.4 -4.613 -5.013 219239_s_at ZNF654 zinc finger protein 654 155.9 140.8 6.8 -4.372 -4.519 220786_s_at SLC38A4 solute carrier family 38, member 4 91.8 135.1 17.7 -2.932 -2.375 206205_at MPHOSPH9 M-phase phosphoprotein 9 208.2 235.7 39.2 -2.588 -2.409 201171_at ATP6V0E ATPase, H+ transporting, lysosomal 9kDa, V0 subunit e 93.5 105 19.1 -2.459 -2.291 204732_s_at TRIM23 tripartite motif-containing 23 132.9 126.3 31 -2.027 -2.100 221079_s_at METTL2B methyltransferase like 2B 141.8 236.9 58.8 -2.010 -1.270 216971_s_at PLEC1 plectin 1, intermediate filament binding protein 500kDa 170 176.3 44.7 -1.980 -1.927 221703_at BRIP1 BRCA1 interacting protein C-terminal helicase 1 137 200.4 52.6 -1.930 -1.381 213647_at DNA2L DNA2 DNA replication helicase 2-like (yeast) 180.5 226.1 61.4 -1.881 -1.556 217547_x_at ZNF675 zinc finger protein 675 89.6 93.3 26 -1.843 -1.785 209865_at SLC35A3 solute carrier family 35 (UDP-N-acetylglucosamine (UDP-GlcNAc) transporter), member A3 191.6 171.3 50.4 -1.765 -1.927 AFFX-M27830_5_atSOX18 SRY (sex determining region Y)-box 18 755.4 982.7 295.8 -1.732 -1.353 212177_at C6orf111 chromosome 6 open reading frame 111 453.6 484.7 147.8 -1.713 -1.618 211088_s_at PLK4 polo-like kinase 4 (Drosophila) 174.6 122.1 37.4 -1.707 -2.223 213880_at LGR5 leucine-rich repeat-containing G protein-coupled receptor 5 172 178 56.2 -1.663 -1.614 201534_s_at -
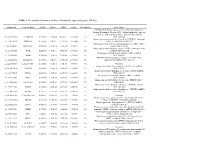
Table 1 the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table 1 The statistical metrics for key differentially expressed genes (DEGs) Agiliant Id Gene Symbol logFC pValue FDR tvalue Regulation Gene Name PREDICTED: Homo sapiens similar to Filamin-C (Gamma- filamin) (Filamin-2) (Protein FLNc) (Actin-binding-like protein) (ABP-L) (ABP-280-like protein) (LOC649056), mRNA A_24_P237896 LOC649056 0.509843 9.18E-14 4.54E-11 12.07302 Up [XR_018580] Homo sapiens programmed cell death 10 (PDCD10), transcript A_23_P18325 PDCD10 0.243111 2.8E-12 9.24E-10 10.62808 Up variant 1, mRNA [NM_007217] Homo sapiens Rho GTPase activating protein 27 (ARHGAP27), A_23_P141335 ARHGAP27 0.492709 3.97E-12 1.22E-09 10.48571 Up mRNA [NM_199282] Homo sapiens tubby homolog (mouse) (TUB), transcript variant A_23_P53110 TUB 0.528219 1.77E-11 4.56E-09 9.891033 Up 1, mRNA [NM_003320] Homo sapiens MyoD family inhibitor (MDFI), mRNA A_23_P42168 MDFI 0.314474 1.81E-10 3.74E-08 8.998697 Up [NM_005586] PREDICTED: Homo sapiens hypothetical LOC644701 A_32_P56890 LOC644701 0.444703 3.6E-10 7.09E-08 8.743973 Up (LOC644701), mRNA [XM_932316] A_32_P167111 A_32_P167111 0.873588 7.41E-10 1.4E-07 8.47781 Up Unknown Homo sapiens zinc finger protein 784 (ZNF784), mRNA A_24_P221424 ZNF784 0.686781 9.18E-10 1.68E-07 8.399687 Up [NM_203374] Homo sapiens lin-28 homolog (C. elegans) (LIN28), mRNA A_23_P74895 LIN28 0.218876 1.27E-09 2.24E-07 8.282224 Up [NM_024674] Homo sapiens ribosomal protein L5 (RPL5), mRNA A_23_P12140 RPL5 0.247598 1.81E-09 3.11E-07 8.154317 Up [NM_000969] Homo sapiens cDNA FLJ43841 fis, clone TESTI4006137. -
Prediction Key Genes and the Potential Molecular Mechanism for Early Recurrent Pregnancy Loss by Multi-Omics Analysis
Prediction key genes and the potential molecular mechanism for early recurrent pregnancy loss by Multi-Omics analysis Le Huang Guangxi Medical University First Aliated Hospital https://orcid.org/0000-0002-1450-2090 Changqiang Wei Guangxi Medical University First Aliated Hospital Zhao Zhang Guangxi Medical University First Aliated Hospital Jiao Cen Guangxi Medical University First Aliated Hospital Zhiwei Zhu Nanjing University of Chinese Medicine Lingjie Deng Guangxi Medical University First Aliated Hospital Lihong Pang ( [email protected] ) https://orcid.org/0000-0002-5179-8396 Research article Keywords: Early recurrent pregnancy loss, Multi-omics analysis, Methylated differentially expressed genes, Transcription factors Posted Date: February 28th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-15435/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/16 Abstract Background: The potential mechanism of early recurrent pregnancy loss (ERPL) has not been fully elucidated, a multi-omics analysis can help us nd key genes. Results: We download data from Gene Expression Omnibus (GEO), 90 hypermethylation-down regulated genes and 49 hypomethylation-up regulated genes were identied by intersection. Compared with the normal early pregnancy group, the expression of ERCC2 and MLH1 was lower in ERPL group, but the expression of PLEK and FOS was higher, and the expression of MLH1 was statistically signicant (p<0.05). Compared with the anembryonic (empty sac) miscarriage group, the expression of MLH1, PLEK, FOS decreased, and that of ERCC2 increased in the embryonic miscarriage group. TF-MDEGs networks predicted SP1, POLR2A, YY1, CREM and CREB1 were involved in methylation regulation of DNA promoter with MDEGs. -

Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription
Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Xiao, Rui et al. "Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription." Cell 178, 1 (June 2019): P107-121.e18 © 2019 Elsevier Inc As Published http://dx.doi.org/10.1016/j.cell.2019.06.001 Publisher Elsevier BV Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/126119 Terms of Use Creative Commons Attribution-NonCommercial-NoDerivs License Detailed Terms http://creativecommons.org/licenses/by-nc-nd/4.0/ HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Cell. Author Manuscript Author manuscript; Manuscript Author available in PMC 2019 September 25. Published in final edited form as: Cell. 2019 June 27; 178(1): 107–121.e18. doi:10.1016/j.cell.2019.06.001. Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription Rui Xiao1,2,11,*, Jia-Yu Chen1,11, Zhengyu Liang1,3,11, Daji Luo1,4, Geng Chen5, Zhi John Lu3, Yang Chen3, Bing Zhou1, Hairi Li1, Xian Du5, Yang Yang5, Mingkui San2, Xintao Wei6, Wen Liu7, Eric Lécuyer8, Brenton R. Graveley6, Gene W. Yeo1, Christopher B. Burge9, Michael Q. Zhang3,10, Yu Zhou5, Xiang-Dong Fu1,12,* 1Department of Cellular and Molecular Medicine, Institute of Genomic Medicine, University of California, San Diego, La Jolla, CA 92093, USA 2Medical Research Institute,