Adaptive Tolerance to a Pathogenic Fungus Drives Major
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Directional Selection and Variation in Finite Populations
Copyright 0 1987 by the Genetics Society of America Directional Selection and Variation in Finite Populations Peter D. Keightley and ‘William G. Hill Institute of Animal Genetics, University of Edinburgh, West Mains Road, Edinburgh EH9 3JN, Scotland Manuscript received April 12, 1987 Revised copy accepted July 16, 1987 ABSTRACT Predictions are made of the equilibrium genetic variances and responses in a metric trait under the joint effects of directional selection, mutation and linkage in a finite population. The “infinitesimal model“ is analyzed as the limiting case of many mutants of very small effect, otherwise Monte Carlo simulation is used. If the effects of mutant genes on the trait are symmetrically distributed and they are unlinked, the variance of mutant effects is not an important parameter. If the distribution is skewed, unless effects or the population size is small, the proportion of mutants that have increasing effect is the critical parameter. With linkage the distribution of genotypic values in the population becomes skewed downward and the equilibrium genetic variance and response are smaller as disequilibrium becomes important. Linkage effects are greater when the mutational variance is contributed by many genes of small effect than few of large effect, and are greater when the majority of mutants increase rather than decrease the trait because genes that are of large effect or are deleterious do not segregate for long. The most likely conditions for ‘‘Muller’s ratchet” are investi- gated. N recent years there has been much interest in the is attained more quickly in the presence of selection I production and maintenance of variation in pop- than in its absence, and is highly dependent on popu- ulations by mutation, stimulated by the presence of lation size. -

Patterns and Power of Phenotypic Selection in Nature
Articles Patterns and Power of Phenotypic Selection in Nature JOEL G. KINGSOLVER AND DAVID W. PFENNIG Phenotypic selection occurs when individuals with certain characteristics produce more surviving offspring than individuals with other characteristics. Although selection is regarded as the chief engine of evolutionary change, scientists have only recently begun to measure its action in the wild. These studies raise numerous questions: How strong is selection, and do different types of traits experience different patterns of selection? Is selection on traits that affect mating success as strong as selection on traits that affect survival? Does selection tend to favor larger body size, and, if so, what are its consequences? We explore these questions and discuss the pitfalls and future prospects of measuring selection in natural populations. Keywords: adaptive landscape, Cope’s rule, natural selection, rapid evolution, sexual selection henotypic selection occurs when individuals with selection on traits that affect survival stronger than on those Pdifferent characteristics (i.e., different phenotypes) that affect only mating success? In this article, we explore these differ in their survival, fecundity, or mating success. The idea and other questions about the patterns and power of phe- of phenotypic selection traces back to Darwin and Wallace notypic selection in nature. (1858), and selection is widely accepted as the primary cause of adaptive evolution within natural populations.Yet Darwin What is selection, and how does it work? never attempted to measure selection in nature, and in the Selection is the nonrandom differential survival or repro- century following the publication of On the Origin of Species duction of phenotypically different individuals. -

Selection and Gene Duplication: a View from the Genome Comment Andreas Wagner
http://genomebiology.com/2002/3/5/reviews/1012.1 Minireview Selection and gene duplication: a view from the genome comment Andreas Wagner Address: Department of Biology, University of New Mexico, 167A Castetter Hall, Albuquerque, NM 817131-1091, USA. E-mail: [email protected] Published: 15 April 2002 Genome Biology 2002, 3(5):reviews1012.1–1012.3 reviews The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2002/3/5/reviews/1012 © BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) Abstract reports Immediately after a gene duplication event, the duplicate genes have redundant functions. Is natural selection therefore completely relaxed after duplication? Does one gene evolve more rapidly than the other? Several recent genome-wide studies have suggested that duplicate genes are always under purifying selection and do not always evolve at the same rate. deposited research When a gene duplication event occurs, the duplicate genes synonymous (silent) nucleotide substitutions, Ks, and have redundant functions. Many deleterious mutations may second, of non-synonymous nucleotide substitutions (which then be harmless, because even if one gene suffers a muta- change the encoded amino acid), Ka (see Box 1). The ratio tion, the redundant gene copy can provide a back-up func- Ka/Ks provides a measure of the selection pressure to which tion. Put differently, after gene duplication - which can arise a gene pair is subject. If a duplicate gene pair shows a Ka/Ks through polyploidization (whole-genome duplication), non- ratio of about 1, that is, if amino-acid replacement substitu- homologous recombination, or through the action of retro- tions occur at the same rate as synonymous substitutions, transposons - one or both duplicates should experience then few or no amino-acid replacement substitutions have refereed research relaxed selective constraints that result in elevated rates of been eliminated since the gene duplication. -
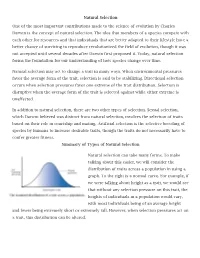
Natural Selection One of the Most Important Contributions Made to The
Natural Selection One of the most important contributions made to the science of evolution by Charles Darwin is the concept of natural selection. The idea that members of a species compete with each other for resources and that individuals that are better adapted to their lifestyle have a better chance of surviving to reproduce revolutionized the field of evolution, though it was not accepted until several decades after Darwin first proposed it. Today, natural selection forms the foundation for our understanding of how species change over time. Natural selection may act to change a trait in many ways. When environmental pressures favor the average form of the trait, selection is said to be stabilizing. Directional selection occurs when selection pressures favor one extreme of the trait distribution. Selection is disruptive when the average form of the trait is selected against while either extreme is unaffected. In addition to natural selection, there are two other types of selection. Sexual selection, which Darwin believed was distinct from natural selection, involves the selection of traits based on their role in courtship and mating. Artificial selection is the selective breeding of species by humans to increase desirable traits, though the traits do not necessarily have to confer greater fitness. Summary of Types of Natural Selection Natural selection can take many forms. To make talking about this easier, we will consider the distribution of traits across a population in using a graph. To the right is a normal curve. For example, if we were talking about height as a trait, we would see that without any selection pressure on this trait, the heights of individuals in a population would vary, with most individuals being of an average height and fewer being extremely short or extremely tall. -

Detecting the Form of Selection from DNA Sequence Data
Outlook COMMENT Male:female mutation ratio muscular dystrophy, neurofibromatosis and retinoblast- outcome of further investigation, this unique transposition oma), there is a large male excess. So, I believe the evidence event and perhaps others like it are sure to provide impor- is convincing that the male base-substitution rate greatly tant cytogenetic and evolutionary insights. exceeds the female rate. There are a number of reasons why the historical Acknowledgements male:female mutation ratio is likely to be less than the I have profited greatly from discussions with my colleagues current one, the most important being the lower age of B. Dove, B. Engels, C. Denniston and M. Susman. My reproduction in the earliest humans and their ape-like greatest debt is to D. Page for a continuing and very fruitful ancestors. More studies are needed. But whatever the dialog and for providing me with unpublished data. References Nature 406, 622–625 6 Engels, W.R. et al. (1994) Long-range cis preference in DNA 1 Vogel, F. and Motulsky, A. G. (1997) Human Genetics: 4 Shimmin, L.C. et al. (1993) Potential problems in estimating homology search over the length of a Drosophila chromosome. Problems and Approaches. Springer-Verlag the male-to-female mutation rate ratio from DNA sequence Science 263, 1623–1625 2 Crow, J. F. (1997) The high spontaneous mutation rate: is it a data. J. Mol. Evol. 37, 160–166 7 Crow, J.F. (2000) The origins, patterns and implications of health risk? Proc. Natl. Acad. Sci. U. S. A. 94, 8380–8386 5 Richardson, C. et al. -

The Peppered Moth and Industrial Melanism: Evolution of a Natural Selection Case Study
Heredity (2013) 110, 207–212 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy REVIEW The peppered moth and industrial melanism: evolution of a natural selection case study LM Cook1 and IJ Saccheri2 From the outset multiple causes have been suggested for changes in melanic gene frequency in the peppered moth Biston betularia and other industrial melanic moths. These have included higher intrinsic fitness of melanic forms and selective predation for camouflage. The possible existence and origin of heterozygote advantage has been debated. From the 1950s, as a result of experimental evidence, selective predation became the favoured explanation and is undoubtedly the major factor driving the frequency change. However, modelling and monitoring of declining melanic frequencies since the 1970s indicate either that migration rates are much higher than existing direct estimates suggested or else, or in addition, non-visual selection has a role. Recent molecular work on genetics has revealed that the melanic (carbonaria) allele had a single origin in Britain, and that the locus is orthologous to a major wing patterning locus in Heliconius butterflies. New methods of analysis should supply further information on the melanic system and on migration that will complete our understanding of this important example of rapid evolution. Heredity (2013) 110, 207–212; doi:10.1038/hdy.2012.92; published online 5 December 2012 Keywords: Biston betularia; carbonaria gene; mutation; predation; non-visual selection; migration INTRODUCTION EARLY EVIDENCE OF CHANGE The peppered moth Biston betularia (L.) and its melanic mutant will The peppered moth was the most diagrammatic example of the be familiar to readers of Heredity as an example of rapid evolutionary phenomenon of industrial melanism that came to be recognised in change brought about by natural selection in a changing environment, industrial and smoke-blackened parts of England in the mid-nine- evenifthedetailsofthestoryarenot.Infact,thedetailsarelesssimple teenth century. -

Natural Selection in a Contemporary Human Population
Natural selection in a contemporary human population Sean G. Byarsa, Douglas Ewbankb, Diddahally R. Govindarajuc, and Stephen C. Stearnsa,1 aDepartment of Ecology and Evolutionary Biology, Yale University, New Haven, CT 06520-8102; bPopulation Studies Center, University of Pennsylvania, Philadelphia, PA 19104-6299; and cDepartment of Neurology, Boston University School of Medicine, Boston, MA 02118-2526 Edited by Peter T. Ellison, Harvard University, Cambridge, MA, and approved September 16, 2009 (received for review June 25, 2009) Our aims were to demonstrate that natural selection is operating sity to identify factors that contribute to cardiovascular disease. on contemporary humans, predict future evolutionary change for It is the longest running multigenerational study in medical specific traits with medical significance, and show that for some history. The people originally enrolled in the study were of pre- traits we can make short-term predictions about our future evolu- dominantly European ancestry (20% United Kingdom, 40% tion. To do so, we measured the strength of selection, estimated Ireland, 10% Italy, 10% Quebec). The original cohort (n = genetic variation and covariation, and predicted the response to 5,209) has been examined every 2 years, a total of 29 times selection for women in the Framingham Heart Study, a project of between 1948 and 2008. The offspring cohort (n = 5,124) has the National Heart, Lung, and Blood Institute and Boston Univer- been examined approximately every 4 years, a total of eight times sity that began in 1948. We found that natural selection is acting between 1971 and 2008 (4). There is also a third generation to cause slow, gradual evolutionary change. -

Major Histocompatibility Complex Selection Dynamics in Pathogen
Downloaded from http://rsbl.royalsocietypublishing.org/ on August 17, 2016 Evolutionary biology Major histocompatibility complex rsbl.royalsocietypublishing.org selection dynamics in pathogen-infected tu´ngara frog (Physalaemus pustulosus) populations Research 1,† 1,† 1 1 Cite this article: Kosch TA, Bataille A, Tiffany A. Kosch , Arnaud Bataille , Chelsea Didinger , John A. Eimes , Didinger C, Eimes JA, Rodrı´guez-Brenes S, Ryan Sofia Rodrı´guez-Brenes2, Michael J. Ryan2,3 and Bruce Waldman1 MJ, Waldman B. 2016 Major histocompatibility 1 complex selection dynamics in pathogen- Laboratory of Behavioral and Population Ecology, School of Biological Sciences, Seoul National University, Seoul 08826, South Korea infected tu´ngara frog (Physalaemus pustulosus) 2Department of Integrative Biology, University of Texas, Austin, TX 78712, USA populations. Biol. Lett. 12: 20160345. 3Smithsonian Tropical Research Institute, Balboa, Anco´n, Panama http://dx.doi.org/10.1098/rsbl.2016.0345 BW, 0000-0003-0006-5333 Pathogen-driven selection can favour major histocompatibility complex (MHC) alleles that confer immunological resistance to specific diseases. How- Received: 25 April 2016 ever, strong directional selection should deplete genetic variation necessary for Accepted: 25 July 2016 robust immune function in the absence of balancing selection or challenges presented by other pathogens. We examined selection dynamics at one MHC class II (MHC-II) locus across Panamanian populations of the tu´ngara frog, Physalaemus pustulosus, infected by the amphibian chytrid fungus Batrachochytrium dendrobatidis (Bd). We compared MHC-II diversity in high- Subject Areas: land tu´ ngara frog populations, where amphibian communities have ecology, evolution, health and experienced declines owing to Bd, with those in the lowland region that disease and epidemiology have shown no evidence of decline. -

Biol2007 - Evolution of Quantitative Characters
1 BIOL2007 - EVOLUTION OF QUANTITATIVE CHARACTERS How do evolutionary biologists measure variation in a typical quantitative character? Let’s use beak size in birds as a typical example. Phenotypic variation (Vp) in a quantitative character Some of the variation in beak size is caused by environmental factors (VE). Some of the variation is caused by genetic factors (VG). If there was no variation in genotype between birds then individuals would differ in beak size only because of different rearing environments. Quantitative geneticists think in terms of how the value of a character in an individual compares to the mean value of the character in the population. The population mean is taken to be a neutral measure and then particular phenotypes are increases or decreases from that value. AT LEVEL OF INDIVIDUAL Phenotype = genotype + environment P = G + E AT LEVEL OF POPULATION Phenotypic variance = genotypic variance + environmental variance VP = VG + VE Genotypic variance is divided into additive, dominance and interaction components VG = VA + VD + V I VA = Additive variance – due to the additive effects of individual alleles (inherited) VD = Dominance variance - reflect the combinations of alleles at each locus (homo- versus (homozygotes versus hetero-zygotes) in any given generation but are not inherited by offspring. An intra-locus effect. VI = Interaction/epistatic variance - reflect epistatic interactions between alleles at different loci. Again not passed onto offspring; they depend on particular combinations of genes in a given generation and are not inherited by offspring. An inter-locus effect. ___________________________________________________________ The following section (italicised) provides the alebgra to backup my assertion that VD and VI are not heritable. -

Directional Selection Can Drive the Evolution of Modularity in Complex Traits
Directional selection can drive the evolution of modularity in complex traits Diogo Melo1 and Gabriel Marroig Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo, Sao Paulo, SP 05508-090, Brazil Edited by Stevan J. Arnold, Oregon State University, Corvallis, OR, and accepted by the Editorial Board December 2, 2014 (received for review December 9, 2013) Modularity is a central concept in modern biology, providing a promotes changes in only one trait of a module, the other traits powerful framework for the study of living organisms on many within this module will suffer an indirect selection pressure and organizational levels. Two central and related questions can be change as well, even if this response leads to lower fitness (8). This posed in regard to modularity: How does modularity appear in the indirect response in other traits is due to their genetic correlation first place, and what forces are responsible for keeping and/or with the selected trait. Understanding how traits become associated, changing modular patterns? We approached these questions using or correlated, is therefore a central question in evolutionary biology. a quantitative genetics simulation framework, building on pre- The question of how modular patterns evolve in each level of vious results obtained with bivariate systems and extending them complexity is still open to intense scrutiny (9–11). In morpho- to multivariate systems. We developed an individual-based model logical systems, one condition for the evolution of variational capable of simulating many traits controlled by many loci with pattern is the existence of genetic variation in the association variable pleiotropic relations between them, expressed in pop- between traits in a population. -

Treatment of Particular Topics. Thus Markert Illustrates Some of the Done
REVIEWS 333 MENDELCENTENARY: GENETICS, DEVELOPMENT AND EVOLUTION (Proceedings of a Symposium held at the Catholic University of America, November 3rd, 1965). Edited by Roland M. Nardone. The Catholic University of America Press, Washington. pp. 174+v. Thismemorial volume, published rather belatedly nearly three years after the event, contains seven short essays and a translation (the same one as in Bateson's Mendel's Principles of Heredity) of Mendel's famous paper. The essays are mostly aimed at a non-specialist audience, but some of them will be interesting to professional geneticists by virtue of their detailed treatment of particular topics. Thus Markert illustrates some of the problems and ideas concerning the regulation of gene activity in higher organisms by reference to lactate dehydrogenase isozymes and Bolton discusses the determination of homologies between DNA of different species using hybridisation techniques. Both these contributions are profusely illustrated (all the speaker's slides seem to have been reproduced) and are sufficiently detailed to be useful references, though much more has been done on DNA hybridisation since 1965. Among other contributors, Davidson discusses the use of numerical scores based on multiple characters in the disentanglement of difficult taxonomic groups, and Dorothea Bennett uses the Tailless series of alleles in the mouse to show how the analysis of the developmental basis of genetic abnormalities can aid understanding of normal development. Lewontin's essay on the gene and evolution presents ideas rather than information. It contains a thoughtful and thought-provoking assessment of the genetic and physiological advantages of the diploid way of life. Nardone's summary of molecular biology in relation to the origin of life seems, in contrast, rather elementary and second-hand, though it may have been correctly judged for the audience. -

Measuring Selection in Contemporary Human Populations
Nature Reviews Genetics | AOP, published online 3 August 2010; doi:10.1038/nrg2831 REVIEWS Measuring selection in contemporary human populations Stephen C. Stearns*, Sean G. Byars*‡, Diddahally R. Govindaraju§ and Douglas Ewbank|| Abstract | Are humans currently evolving? This question can be answered using data on lifetime reproductive success, multiple traits and genetic variation and covariation in those traits. Such data are available in existing long-term, multigeneration studies — both clinical and epidemiological — but they have not yet been widely used to address contemporary human evolution. Here we review methods to predict evolutionary change and attempts to measure selection and inheritance in humans. We also assemble examples of long-term studies in which additional measurements of evolution could be made. The evidence strongly suggests that we are evolving and that our nature is dynamic, not static. Given the recent emphasis on the roles of genes in research because of the insights it offers into short-term disease and evolution, we tend to forget that in the short changes in the health and demography of human popu- run evolution is driven by differences in fitness among lations. Research on evolution in contemporary human phenotypes, not genotypes. We can learn a lot about the populations could also change our understanding of the speed and direction of evolution in humans by exam- long-term implications of recent medical innovations. ining the transmission of phenotypes from one gen- Medical practice and publicity about its results affect the eration to the next. This focus on phenotypes contrasts direction and intensity of selection on traits of medical with the attention recently paid to the signatures that interest.