Molecular Mechanisms of Human Γδ T Cell Activation and Tumor Cell Recognition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

List of Genes Used in Cell Type Enrichment Analysis
List of genes used in cell type enrichment analysis Metagene Cell type Immunity ADAM28 Activated B cell Adaptive CD180 Activated B cell Adaptive CD79B Activated B cell Adaptive BLK Activated B cell Adaptive CD19 Activated B cell Adaptive MS4A1 Activated B cell Adaptive TNFRSF17 Activated B cell Adaptive IGHM Activated B cell Adaptive GNG7 Activated B cell Adaptive MICAL3 Activated B cell Adaptive SPIB Activated B cell Adaptive HLA-DOB Activated B cell Adaptive IGKC Activated B cell Adaptive PNOC Activated B cell Adaptive FCRL2 Activated B cell Adaptive BACH2 Activated B cell Adaptive CR2 Activated B cell Adaptive TCL1A Activated B cell Adaptive AKNA Activated B cell Adaptive ARHGAP25 Activated B cell Adaptive CCL21 Activated B cell Adaptive CD27 Activated B cell Adaptive CD38 Activated B cell Adaptive CLEC17A Activated B cell Adaptive CLEC9A Activated B cell Adaptive CLECL1 Activated B cell Adaptive AIM2 Activated CD4 T cell Adaptive BIRC3 Activated CD4 T cell Adaptive BRIP1 Activated CD4 T cell Adaptive CCL20 Activated CD4 T cell Adaptive CCL4 Activated CD4 T cell Adaptive CCL5 Activated CD4 T cell Adaptive CCNB1 Activated CD4 T cell Adaptive CCR7 Activated CD4 T cell Adaptive DUSP2 Activated CD4 T cell Adaptive ESCO2 Activated CD4 T cell Adaptive ETS1 Activated CD4 T cell Adaptive EXO1 Activated CD4 T cell Adaptive EXOC6 Activated CD4 T cell Adaptive IARS Activated CD4 T cell Adaptive ITK Activated CD4 T cell Adaptive KIF11 Activated CD4 T cell Adaptive KNTC1 Activated CD4 T cell Adaptive NUF2 Activated CD4 T cell Adaptive PRC1 Activated -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

Focus on Γδ T and NK Cells
cells Review Engineering the Bridge between Innate and Adaptive Immunity for Cancer Immunotherapy: Focus on γδ T and NK Cells 1, 2, 1 2, , Fabio Morandi y , Mahboubeh Yazdanifar y , Claudia Cocco , Alice Bertaina * z and 1, , Irma Airoldi * z 1 Stem Cell Laboratory and Cell Therapy Center, IRCCS Istituto Giannina Gaslini, Via G. Gaslini, 516147 Genova, Italy; [email protected] (F.M.); [email protected] (C.C.) 2 Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford University School of Medicine, Palo Alto, CA 94305, USA; [email protected] * Correspondence: [email protected] (A.B.); [email protected] (I.A.) These authors have contributed equally to this work as first author. y These authors have contributed equally to this work as last author. z Received: 1 June 2020; Accepted: 21 July 2020; Published: 22 July 2020 Abstract: Most studies on genetic engineering technologies for cancer immunotherapy based on allogeneic donors have focused on adaptive immunity. However, the main limitation of such approaches is that they can lead to severe graft-versus-host disease (GvHD). An alternative approach would bolster innate immunity by relying on the natural tropism of some subsets of the innate immune system, such as γδ T and natural killer (NK) cells, for the tumor microenvironment and their ability to kill in a major histocompatibility complex (MHC)-independent manner. γδ T and NK cells have the unique ability to bridge innate and adaptive immunity while responding to a broad range of tumors. Considering these properties, γδ T and NK cells represent ideal sources for developing allogeneic cell therapies. -

Pan-KIR2DL NK-Receptor Antibodies and Their Use in Diagnostik and Therapy
(19) TZZ _ T (11) EP 2 287 195 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.02.2011 Bulletin 2011/08 C07K 16/28 (2006.01) G01N 33/52 (2006.01) (21) Application number: 10178924.6 (22) Date of filing: 01.07.2005 (84) Designated Contracting States: • Romagne, François AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 13600 La Ciotat (FR) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Wagtmann, Peter, Andreas, Nicolai, Reumert SK TR 2960 Rungsted Kyst (DK) • Svendsen, Ivan (30) Priority: 06.01.2005 DK 200500025 2765 Smorum (DK) 01.07.2004 PCT/DK2004/000470 • Zahn, Stefan 01.07.2004 PCT/IB2004/002464 2750 Ballerup (DK) • Svensson, Anders (62) Document number(s) of the earlier application(s) in 21746 Malmö (DK) accordance with Art. 76 EPC: • Thorolfsson, Matthias 05758642.2 / 1 791 868 2920 Charlottenlund (DK) • Berg Padkaer, Soren (71) Applicants: 3500 Vaerlose (DK) • Novo Nordisk A/S • Kjaergaard, Kristian 2880 Bagsvaerd (DK) 2750 Ballerup (DK) • Innate Pharma • Spee, Pieter 13009 Marseille (FR) 3450 Allerod (DK) • Universita di Genova • Wilken, Michael 16132 Genova (IT) 3390 Hundested (DK) (72) Inventors: (74) Representative: Gallois, Valérie et al • Moretta, Alessandro Cabinet BECKER & ASSOCIES 16133 Genova (IT) 25, rue Louis Le Grand • Della Chiesa, Mariella 75002 Paris (FR) 16132 Genova (IT) • Andre, Pascale Remarks: 13006 Marseille (FR) This application was filed on 23-09-2010 as a • Gauthier, Laurent divisional application to the application mentioned 13008 Marseille (FR) under INID code 62. -

Predominant Activation and Expansion of V Gamma 9-Bearing Gamma Delta T Cells in Vivo As Well As in Vitro in Salmonella Infection
Predominant activation and expansion of V gamma 9-bearing gamma delta T cells in vivo as well as in vitro in Salmonella infection. T Hara, … , G Matsuzaki, Y Yoshikai J Clin Invest. 1992;90(1):204-210. https://doi.org/10.1172/JCI115837. Research Article Gamma delta T cell receptor-positive cells (gamma delta T cells) have recently been implicated to play a role in the protection against infectious pathogens. Serial studies on gamma delta T cells in 14 patients with salmonella infection have revealed that the proportions of gamma delta T cells (mean +/- SD: 17.9 +/- 13.2%) in salmonella infection were significantly increased (P less than 0.01) compared with 35 normal controls (5.0 +/- 2.6%) and 13 patients with other bacterial infections (4.0 +/- 1.4%). Expansion of gamma delta T cells was more prominent in the systemic form (28.9 +/- 10.8%) than in the gastroenteritis form (10.5 +/- 7.9%) of salmonella infection (P less than 0.01). Most in vivo-expanded gamma delta T cells expressed V gamma 9 gene product. Increased activated (HLA-DR+) T cells were observed in all the six patients with the systemic form and four of the seven with gastroenteritis form. Especially in the six with systemic form, gamma delta T cell activation was significantly higher than alpha beta T cell activation at the early stage of illness (P less than 0.01). When peripheral blood lymphocytes from normal individuals were cultured with live salmonella, gamma delta T cells were preferentially activated and expanded and most of them expressed V gamma 9. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Borne, Richard (2019) Natural Killer (NK) Cell Receptor Expression in Cattle: Development of a Pipeline for Accurate Quantification of RNA Sequencing (RNA-Seq) Data
Borne, Richard (2019) Natural Killer (NK) cell receptor expression in cattle: development of a pipeline for accurate quantification of RNA Sequencing (RNA-Seq) data. PhD thesis. https://theses.gla.ac.uk/71955/ Copyright and moral rights for this work are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This work cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Enlighten: Theses https://theses.gla.ac.uk/ [email protected] Natural Killer (NK) cell receptor expression in cattle: development of a pipeline for accurate quantification of RNA Sequencing (RNA-Seq) data Richard Borne BSc. (Hons) Submitted in fulfilment of the requirements for the Degree of PhD College of Medical, Veterinary & Life Sciences University of Glasgow September 2018 2 Abstract Cattle have undergone significant gene expansion within two of the major NK receptor encoding complexes, the leukocyte receptor complex (LRC) and natural killer complex (NKC). This expansion has resulted in a number of highly similar genes densely packed within each complex. The genes can be highly polymorphic and encode for fundamentally important receptors for controlling the functional response of NK cells. Understanding the nature of transcription of the LRC and NKC genes is required to confirm existing gene models, predicted functional status and also their distribution in cell and tissue types. -

Gene List HTG Edgeseq Immuno-Oncology Assay
Gene List HTG EdgeSeq Immuno-Oncology Assay Adhesion ADGRE5 CLEC4A CLEC7A IBSP ICAM4 ITGA5 ITGB1 L1CAM MBL2 SELE ALCAM CLEC4C DST ICAM1 ITGA1 ITGA6 ITGB2 LGALS1 MUC1 SVIL CDH1 CLEC5A EPCAM ICAM2 ITGA2 ITGAL ITGB3 LGALS3 NCAM1 THBS1 CDH5 CLEC6A FN1 ICAM3 ITGA4 ITGAM ITGB4 LGALS9 PVR THY1 Apoptosis APAF1 BCL2 BID CARD11 CASP10 CASP8 FADD NOD1 SSX1 TP53 TRAF3 BCL10 BCL2L1 BIRC5 CASP1 CASP3 DDX58 NLRP3 NOD2 TIMP1 TRAF2 TRAF6 B-Cell Function BLNK BTLA CD22 CD79A FAS FCER2 IKBKG PAX5 SLAMF1 SLAMF7 SPN BTK CD19 CD24 EBF4 FASLG IKBKB MS4A1 RAG1 SLAMF6 SPI1 Cell Cycle ABL1 ATF1 ATM BATF CCND1 CDK1 CDKN1B NCL RELA SSX1 TBX21 TP53 ABL2 ATF2 AXL BAX CCND3 CDKN1A EGR1 REL RELB TBK1 TIMP1 TTK Cell Signaling ADORA2A DUSP4 HES1 IGF2R LYN MAPK1 MUC1 NOTCH1 RIPK2 SMAD3 STAT5B AKT3 DUSP6 HES5 IKZF1 MAF MAPK11 MYC PIK3CD RNF4 SOCS1 STAT6 BCL6 ELK1 HEY1 IKZF2 MAP2K1 MAPK14 NFATC1 PIK3CG RORC SOCS3 SYK CEBPB EP300 HEY2 IKZF3 MAP2K2 MAPK3 NFATC3 POU2F2 RUNX1 SPINK5 TAL1 CIITA ETS1 HEYL JAK1 MAP2K4 MAPK8 NFATC4 PRKCD RUNX3 STAT1 TCF7 CREB1 FLT3 HMGB1 JAK2 MAP2K7 MAPKAPK2 NFKB1 PRKCE S100B STAT2 TYK2 CREB5 FOS HRAS JAK3 MAP3K1 MEF2C NFKB2 PTEN SEMA4D STAT3 CREBBP GATA3 IGF1R KIT MAP3K5 MTDH NFKBIA PYCARD SMAD2 STAT4 Chemokine CCL1 CCL16 CCL20 CCL25 CCL4 CCR2 CCR7 CX3CL1 CXCL12 CXCL3 CXCR1 CXCR6 CCL11 CCL17 CCL21 CCL26 CCL5 CCR3 CCR9 CX3CR1 CXCL13 CXCL5 CXCR2 MST1R CCL13 CCL18 CCL22 CCL27 CCL7 CCR4 CCRL2 CXCL1 CXCL14 CXCL6 CXCR3 PPBP CCL14 CCL19 CCL23 CCL28 CCL8 CCR5 CKLF CXCL10 CXCL16 CXCL8 CXCR4 XCL2 CCL15 CCL2 CCL24 CCL3 CCR1 CCR6 CMKLR1 CXCL11 CXCL2 CXCL9 CXCR5 -

T Cells Via IL-2 Production Δγ Human
B7−CD28 Costimulatory Signals Control the Survival and Proliferation of Murine and Human δγ T Cells via IL-2 Production This information is current as Julie C. Ribot, Ana deBarros, Liliana Mancio-Silva, Ana of September 24, 2021. Pamplona and Bruno Silva-Santos J Immunol 2012; 189:1202-1208; Prepublished online 25 June 2012; doi: 10.4049/jimmunol.1200268 http://www.jimmunol.org/content/189/3/1202 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2012/06/25/jimmunol.120026 Material 8.DC1 http://www.jimmunol.org/ References This article cites 49 articles, 18 of which you can access for free at: http://www.jimmunol.org/content/189/3/1202.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 24, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology B7–CD28 Costimulatory Signals Control the Survival and Proliferation of Murine and Human gd T Cells via IL-2 Production Julie C. -

Human Peripheral Blood Gamma Delta T Cells: Report on a Series of Healthy Caucasian Portuguese Adults and Comprehensive Review of the Literature
cells Article Human Peripheral Blood Gamma Delta T Cells: Report on a Series of Healthy Caucasian Portuguese Adults and Comprehensive Review of the Literature 1, 2, 1, 1, Sónia Fonseca y, Vanessa Pereira y, Catarina Lau z, Maria dos Anjos Teixeira z, Marika Bini-Antunes 3 and Margarida Lima 1,* 1 Laboratory of Cytometry, Unit for Hematology Diagnosis, Department of Hematology, Hospital de Santo António (HSA), Centro Hospitalar Universitário do Porto (CHUP), Unidade Multidisciplinar de Investigação Biomédica, Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto (UMIB/ICBAS/UP), 4099-001 Porto Porto, Portugal; [email protected] (S.F.); [email protected] (C.L.); [email protected] (M.d.A.T.) 2 Department of Clinical Pathology, Centro Hospitalar de Vila Nova de Gaia/Espinho (CHVNG/E), 4434-502 Vila Nova de Gaia, Portugal; [email protected] 3 Laboratory of Immunohematology and Blood Donors Unit, Department of Hematology, Hospital de Santo António (HSA), Centro Hospitalar Universitário do Porto (CHUP), Unidade Multidisciplinar de Investigação Biomédica, Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto (UMIB/ICBAS/UP), 4099-001Porto, Portugal; [email protected] * Correspondence: [email protected]; Tel.: + 351-22-20-77-500 These authors contributed equally to this work. y These authors contributed equally to this work. z Received: 10 February 2020; Accepted: 13 March 2020; Published: 16 March 2020 Abstract: Gamma delta T cells (Tc) are divided according to the type of Vδ and Vγ chains they express, with two major γδ Tc subsets being recognized in humans: Vδ2Vγ9 and Vδ1. -
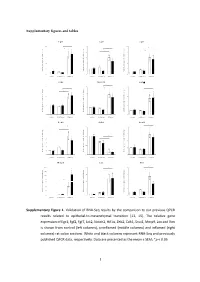
Type of the Paper (Article
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1.