Borne, Richard (2019) Natural Killer (NK) Cell Receptor Expression in Cattle: Development of a Pipeline for Accurate Quantification of RNA Sequencing (RNA-Seq) Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

List of Genes Used in Cell Type Enrichment Analysis
List of genes used in cell type enrichment analysis Metagene Cell type Immunity ADAM28 Activated B cell Adaptive CD180 Activated B cell Adaptive CD79B Activated B cell Adaptive BLK Activated B cell Adaptive CD19 Activated B cell Adaptive MS4A1 Activated B cell Adaptive TNFRSF17 Activated B cell Adaptive IGHM Activated B cell Adaptive GNG7 Activated B cell Adaptive MICAL3 Activated B cell Adaptive SPIB Activated B cell Adaptive HLA-DOB Activated B cell Adaptive IGKC Activated B cell Adaptive PNOC Activated B cell Adaptive FCRL2 Activated B cell Adaptive BACH2 Activated B cell Adaptive CR2 Activated B cell Adaptive TCL1A Activated B cell Adaptive AKNA Activated B cell Adaptive ARHGAP25 Activated B cell Adaptive CCL21 Activated B cell Adaptive CD27 Activated B cell Adaptive CD38 Activated B cell Adaptive CLEC17A Activated B cell Adaptive CLEC9A Activated B cell Adaptive CLECL1 Activated B cell Adaptive AIM2 Activated CD4 T cell Adaptive BIRC3 Activated CD4 T cell Adaptive BRIP1 Activated CD4 T cell Adaptive CCL20 Activated CD4 T cell Adaptive CCL4 Activated CD4 T cell Adaptive CCL5 Activated CD4 T cell Adaptive CCNB1 Activated CD4 T cell Adaptive CCR7 Activated CD4 T cell Adaptive DUSP2 Activated CD4 T cell Adaptive ESCO2 Activated CD4 T cell Adaptive ETS1 Activated CD4 T cell Adaptive EXO1 Activated CD4 T cell Adaptive EXOC6 Activated CD4 T cell Adaptive IARS Activated CD4 T cell Adaptive ITK Activated CD4 T cell Adaptive KIF11 Activated CD4 T cell Adaptive KNTC1 Activated CD4 T cell Adaptive NUF2 Activated CD4 T cell Adaptive PRC1 Activated -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

Pan-KIR2DL NK-Receptor Antibodies and Their Use in Diagnostik and Therapy
(19) TZZ _ T (11) EP 2 287 195 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.02.2011 Bulletin 2011/08 C07K 16/28 (2006.01) G01N 33/52 (2006.01) (21) Application number: 10178924.6 (22) Date of filing: 01.07.2005 (84) Designated Contracting States: • Romagne, François AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 13600 La Ciotat (FR) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Wagtmann, Peter, Andreas, Nicolai, Reumert SK TR 2960 Rungsted Kyst (DK) • Svendsen, Ivan (30) Priority: 06.01.2005 DK 200500025 2765 Smorum (DK) 01.07.2004 PCT/DK2004/000470 • Zahn, Stefan 01.07.2004 PCT/IB2004/002464 2750 Ballerup (DK) • Svensson, Anders (62) Document number(s) of the earlier application(s) in 21746 Malmö (DK) accordance with Art. 76 EPC: • Thorolfsson, Matthias 05758642.2 / 1 791 868 2920 Charlottenlund (DK) • Berg Padkaer, Soren (71) Applicants: 3500 Vaerlose (DK) • Novo Nordisk A/S • Kjaergaard, Kristian 2880 Bagsvaerd (DK) 2750 Ballerup (DK) • Innate Pharma • Spee, Pieter 13009 Marseille (FR) 3450 Allerod (DK) • Universita di Genova • Wilken, Michael 16132 Genova (IT) 3390 Hundested (DK) (72) Inventors: (74) Representative: Gallois, Valérie et al • Moretta, Alessandro Cabinet BECKER & ASSOCIES 16133 Genova (IT) 25, rue Louis Le Grand • Della Chiesa, Mariella 75002 Paris (FR) 16132 Genova (IT) • Andre, Pascale Remarks: 13006 Marseille (FR) This application was filed on 23-09-2010 as a • Gauthier, Laurent divisional application to the application mentioned 13008 Marseille (FR) under INID code 62. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Gene List HTG Edgeseq Immuno-Oncology Assay
Gene List HTG EdgeSeq Immuno-Oncology Assay Adhesion ADGRE5 CLEC4A CLEC7A IBSP ICAM4 ITGA5 ITGB1 L1CAM MBL2 SELE ALCAM CLEC4C DST ICAM1 ITGA1 ITGA6 ITGB2 LGALS1 MUC1 SVIL CDH1 CLEC5A EPCAM ICAM2 ITGA2 ITGAL ITGB3 LGALS3 NCAM1 THBS1 CDH5 CLEC6A FN1 ICAM3 ITGA4 ITGAM ITGB4 LGALS9 PVR THY1 Apoptosis APAF1 BCL2 BID CARD11 CASP10 CASP8 FADD NOD1 SSX1 TP53 TRAF3 BCL10 BCL2L1 BIRC5 CASP1 CASP3 DDX58 NLRP3 NOD2 TIMP1 TRAF2 TRAF6 B-Cell Function BLNK BTLA CD22 CD79A FAS FCER2 IKBKG PAX5 SLAMF1 SLAMF7 SPN BTK CD19 CD24 EBF4 FASLG IKBKB MS4A1 RAG1 SLAMF6 SPI1 Cell Cycle ABL1 ATF1 ATM BATF CCND1 CDK1 CDKN1B NCL RELA SSX1 TBX21 TP53 ABL2 ATF2 AXL BAX CCND3 CDKN1A EGR1 REL RELB TBK1 TIMP1 TTK Cell Signaling ADORA2A DUSP4 HES1 IGF2R LYN MAPK1 MUC1 NOTCH1 RIPK2 SMAD3 STAT5B AKT3 DUSP6 HES5 IKZF1 MAF MAPK11 MYC PIK3CD RNF4 SOCS1 STAT6 BCL6 ELK1 HEY1 IKZF2 MAP2K1 MAPK14 NFATC1 PIK3CG RORC SOCS3 SYK CEBPB EP300 HEY2 IKZF3 MAP2K2 MAPK3 NFATC3 POU2F2 RUNX1 SPINK5 TAL1 CIITA ETS1 HEYL JAK1 MAP2K4 MAPK8 NFATC4 PRKCD RUNX3 STAT1 TCF7 CREB1 FLT3 HMGB1 JAK2 MAP2K7 MAPKAPK2 NFKB1 PRKCE S100B STAT2 TYK2 CREB5 FOS HRAS JAK3 MAP3K1 MEF2C NFKB2 PTEN SEMA4D STAT3 CREBBP GATA3 IGF1R KIT MAP3K5 MTDH NFKBIA PYCARD SMAD2 STAT4 Chemokine CCL1 CCL16 CCL20 CCL25 CCL4 CCR2 CCR7 CX3CL1 CXCL12 CXCL3 CXCR1 CXCR6 CCL11 CCL17 CCL21 CCL26 CCL5 CCR3 CCR9 CX3CR1 CXCL13 CXCL5 CXCR2 MST1R CCL13 CCL18 CCL22 CCL27 CCL7 CCR4 CCRL2 CXCL1 CXCL14 CXCL6 CXCR3 PPBP CCL14 CCL19 CCL23 CCL28 CCL8 CCR5 CKLF CXCL10 CXCL16 CXCL8 CXCR4 XCL2 CCL15 CCL2 CCL24 CCL3 CCR1 CCR6 CMKLR1 CXCL11 CXCL2 CXCL9 CXCR5 -
Type of the Paper (Article
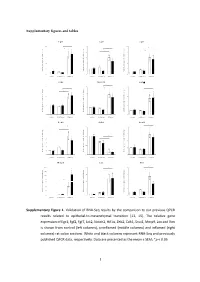
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Systems Analysis of Protective Immune Responses to RTS,S Malaria Vaccination in Humans
Systems analysis of protective immune responses to RTS,S malaria vaccination in humans Dmitri Kazmina,1, Helder I. Nakayab,1, Eva K. Leec, Matthew J. Johnsond, Robbert van der Moste, Robert A. van den Bergf, W. Ripley Ballouf, Erik Jongerte, Ulrike Wille-Reeceg, Christian Ockenhouseg, Alan Aderemh, Daniel E. Zakh, Jerald Sadoffi, Jenny Hendriksi, Jens Wrammerta, Rafi Ahmeda,2, and Bali Pulendrana,j,2 aEmory Vaccine Center, Emory University, Atlanta, GA 30329; bSchool of Pharmaceutical Sciences, University of Sao Paulo, São Paulo 05508, Brazil; cSchool of Industrial and Systems Engineering, Georgia Institute of Technology, Atlanta, GA 30332; dCenter for Genome Engineering, University of Minnesota, Minneapolis, MN 55108; eGSK Vaccines, Rixensart 1330, Belgium; fGSK Vaccines, Rockville, MD 20850; gProgram for Appropriate Technology in Health-Malaria Vaccine Initiative, Washington, DC 20001; hCenter for Infectious Disease Research, Seattle, WA 98109; iCrucell, Leiden 2333, The Netherlands; and jDepartment of Pathology, Emory University School of Medicine, Atlanta, GA 30322 Contributed by Rafi Ahmed, January 4, 2017 (sent for review December 19, 2016; reviewed by Elias Haddad and Robert Seder) RTS,S is an advanced malaria vaccine candidate and confers trials conducted in malaria endemic areas in Africa proved the significant protection against Plasmodium falciparum infection in vaccine to be partially protective in adults (9), children (10, 11), humans. Little is known about the molecular mechanisms driving and infants (12, 13). These results were further confirmed in a vaccine immunity. Here, we applied a systems biology approach phase III trial in sub-Saharan Africa (14–17) in which 55.8% to study immune responses in subjects receiving three consecutive efficacy against clinical malaria was observed over the first 12 mo immunizations with RTS,S (RRR), or in those receiving two immuni- of follow-up in children of 5–17 mo (14). -

Natural Killer Cell Lymphoma Shares Strikingly Similar Molecular Features
Leukemia (2011) 25, 348–358 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu ORIGINAL ARTICLE Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic cd T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro J Iqbal1, DD Weisenburger1, A Chowdhury2, MY Tsai2, G Srivastava3, TC Greiner1, C Kucuk1, K Deffenbacher1, J Vose4, L Smith5, WY Au3, S Nakamura6, M Seto6, J Delabie7, F Berger8, F Loong3, Y-H Ko9, I Sng10, X Liu11, TP Loughran11, J Armitage4 and WC Chan1, for the International Peripheral T-cell Lymphoma Project 1Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 2Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 3Departments of Pathology and Medicine, University of Hong Kong, Queen Mary Hospital, Hong Kong, China; 4Division of Hematology and Oncology, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 5College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA; 6Departments of Pathology and Cancer Genetics, Aichi Cancer Center Research Institute, Nagoya University, Nagoya, Japan; 7Department of Pathology, University of Oslo, Norwegian Radium Hospital, Oslo, Norway; 8Department of Pathology, Centre Hospitalier Lyon-Sud, Lyon, France; 9Department of Pathology, Samsung Medical Center, Sungkyunkwan University, Seoul, Korea; 10Department of Pathology, Singapore General Hospital, Singapore and 11Penn State Hershey Cancer Institute, Pennsylvania State University College of Medicine, Hershey, PA, USA Natural killer (NK) cell lymphomas/leukemias are rare neo- Introduction plasms with an aggressive clinical behavior. -

Understanding the Synergy of Nkp46 and Co-Activating Signals in Various NK Cell Subpopulations: Paving the Way for More Successful NK-Cell-Based Immunotherapy
cells Article Understanding the Synergy of NKp46 and Co-Activating Signals in Various NK Cell Subpopulations: Paving the Way for More Successful NK-Cell-Based Immunotherapy 1,2, 3, 1, 1 Loris Zamai * , Genny Del Zotto y , Flavia Buccella y, Sara Gabrielli , Barbara Canonico 1 , Marco Artico 4, Claudio Ortolani 1 and Stefano Papa 1 1 Department of Biomolecular Sciences, University of Urbino “Carlo Bo”, 61032 Urbino, Italy; fl[email protected] (F.B.); [email protected] (S.G.); [email protected] (B.C.); [email protected] (C.O.); [email protected] (S.P.) 2 INFN-Gran Sasso National Laboratory, Assergi, 67100 L’Aquila, Italy 3 Area Aggregazione Servizi e Laboratori Diagnostici, IRCCS Istituto Giannina Gaslini, 16147 Genoa, Italy; [email protected] 4 Department of Sensory Organs, Sapienza University of Rome, 00161 Rome, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-0722-304319; Fax: +39-0722-304319 These authors contributed equally. y Received: 11 February 2020; Accepted: 13 March 2020; Published: 19 March 2020 Abstract: The NK cell population is characterized by distinct NK cell subsets that respond differently to the various activating stimuli. For this reason, the determination of the optimal cytotoxic activation of the different NK cell subsets can be a crucial aspect to be exploited to counter cancer cells in oncologic patients. To evaluate how the triggering of different combination of activating receptors can affect the cytotoxic responses of different NK cell subsets, we developed a microbead-based degranulation assay. By using this new assay, we were able to detect CD107a+ degranulating NK cells even within the less cytotoxic subsets (i.e., resting CD56bright and unlicensed CD56dim NK cells), thus demonstrating its high sensitivity. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

NK Cells: Receptors and Functions Eric Vivier and Sophie Ugolini
NK cells: receptors and functions Eric Vivier and Sophie Ugolini Natural killer (NK) cells were identified in 1975 as lymphocytes of the innate many processes that were originally restricted to adaptive immunity, such immune system that can kill tumour cells. Since then, NK cells have been as priming, education and memory, are now known to occur in NK cells. shown to kill an array of ‘stressed’ cells and secrete cytokines that Indeed, NK cells undergo sophisticated processes of adaptation that allow participate in shaping adaptive immune responses. A key feature of NK cells them to be tuned to their environment. There is also a growing interest in resides in their capacity to distinguish stressed cells (such as tumour cells, manipulating NK cells in innovative therapeutic settings. For example, the infected cells and damaged cells) from normal cells. Although NK cells are understanding of NK cell inhibition by MHC class I-specific receptors has generally considered to be components of early innate immune defence, prompted the design of innovative anticancer therapies. The NK cell detection system includes numerous receptors, Key receptors on human NK cells the engagement of which dictates the quality and intensity Inhibitory receptors Activating receptors, adhesion molecules or co-stimulatory molecules of the NK cell response. NK cells use inhibitory receptors to gauge the absence of constitutively expressed self molecules Target cell Nectin 2 SLAMF7 Sialic CLEC2D (CD112, PVRL2) NECL2 L-selectin NECL5 AICL NTB-A (CRACC, on susceptible target cells. As a consequence, NK cells can Receptors for MHC class I and class I-Iike molecules B7-H6 acid Cadherins (LLT1) CMV pp65 NECL5 (CADM1) (CD62L) (CD155, PVR) (CLEC2B) CEACAM1 (SLAMF6) CS1, CD319) recognize ‘missing self’ on haematopoietic cells.