Genome-Wide Localization of the RNA Polymerase III Transcription Machinery in Human Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats](https://docslib.b-cdn.net/cover/6136/fk506-binding-protein-12-6-1b-a-negative-regulator-of-ca2-rescues-memory-and-restores-genomic-regulation-in-the-hippocampus-of-aging-rats-16136.webp)
FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats
This Accepted Manuscript has not been copyedited and formatted. The final version may differ from this version. A link to any extended data will be provided when the final version is posted online. Research Articles: Neurobiology of Disease FK506-Binding Protein 12.6/1b, a negative regulator of [Ca2+], rescues memory and restores genomic regulation in the hippocampus of aging rats John C. Gant1, Eric M. Blalock1, Kuey-Chu Chen1, Inga Kadish2, Olivier Thibault1, Nada M. Porter1 and Philip W. Landfield1 1Department of Pharmacology & Nutritional Sciences, University of Kentucky, Lexington, KY 40536 2Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294 DOI: 10.1523/JNEUROSCI.2234-17.2017 Received: 7 August 2017 Revised: 10 October 2017 Accepted: 24 November 2017 Published: 18 December 2017 Author contributions: J.C.G. and P.W.L. designed research; J.C.G., E.M.B., K.-c.C., and I.K. performed research; J.C.G., E.M.B., K.-c.C., I.K., and P.W.L. analyzed data; J.C.G., E.M.B., O.T., N.M.P., and P.W.L. wrote the paper. Conflict of Interest: The authors declare no competing financial interests. NIH grants AG004542, AG033649, AG052050, AG037868 and McAlpine Foundation for Neuroscience Research Corresponding author: Philip W. Landfield, [email protected], Department of Pharmacology & Nutritional Sciences, University of Kentucky, 800 Rose Street, UKMC MS 307, Lexington, KY 40536 Cite as: J. Neurosci ; 10.1523/JNEUROSCI.2234-17.2017 Alerts: Sign up at www.jneurosci.org/cgi/alerts to receive customized email alerts when the fully formatted version of this article is published. -
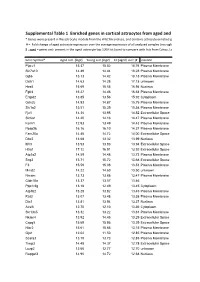
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Nuclear Factor I/B Is an Oncogene in Small Cell Lung Cancer
Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Dooley, A. L. et al. “Nuclear factor I/B is an oncogene in small cell lung cancer.” Genes & Development 25 (2011): 1470-1475. As Published http://dx.doi.org/10.1101/gad.2046711 Publisher Cold Spring Harbor Laboratory Press in association with The Genetics Society Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/66512 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Dooley 1 Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer Alison L. Dooley1, Monte M. Winslow1, Derek Y. Chiang2,3,4, Shantanu Banerji2,3, Nicolas Stransky2, Talya L. Dayton1, Eric L. Snyder1, Stephanie Senna1, Charles A. Whittaker1, Roderick T. Bronson5, Denise Crowley1, Jordi Barretina2,3, Levi Garraway2,3, Matthew Meyerson2,3, Tyler Jacks1,6 1David H. Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2The Broad Institute, Cancer Program, Cambridge, Massachusetts, USA 3Dana-Farber Cancer Institute, Department of Medical Oncology and Center for Cancer Genome Discovery, Boston, Massachusetts, USA 4Current address: Lineberger Comprehensive Cancer Center, 450 West Drive, CB #7295, Chapel Hill, North Carolina, USA 5Department of Pathology, Tufts University School of Medicine and Veterinary Medicine, North Grafton, Massachusetts, USA 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA Key Words: Small Cell Lung Cancer, Mouse model, Nuclear Factor I/B Dooley 2 Abstract Small cell lung cancer (SCLC) is an aggressive cancer often diagnosed after it has metastasized. -

Implication of M6a Mrna Methylation in Susceptibility to Inflammatory
epigenomes Article Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease Maialen Sebastian-delaCruz 1,2 , Ane Olazagoitia-Garmendia 1,2 , Itziar Gonzalez-Moro 2,3 , Izortze Santin 2,3,4 , Koldo Garcia-Etxebarria 5 and Ainara Castellanos-Rubio 1,2,4,6,* 1 Department of Genetics, Physical Anthropology and Animal Fisiology, University of the Basque Country, 48940 Leioa, Spain; [email protected] (M.S.-d.); [email protected] (A.O.-G.) 2 Biocruces Bizkaia Health Research Institute, 48903 Barakaldo, Spain; [email protected] (I.G.-M.); [email protected] (I.S.) 3 Department of Biochemistry and Molecular Biology, University of the Basque Country, 48940 Leioa, Spain 4 CIBER (Centro de Investigación Biomédica en Red) de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), Instituto de Salud Carlos III, 28029 Madrid, Spain 5 Hepatic and Gastrointestinal Disease Area, IIS Biodonostia, 20014 Donostia, Spain; [email protected] 6 Ikerbasque, Basque Foundation for Science, 48013 Bilbao, Spain * Correspondence: [email protected] Received: 29 June 2020; Accepted: 28 July 2020; Published: 3 August 2020 Abstract: Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract that develops due to the interaction between genetic and environmental factors. More than 160 loci have been associated with IBD, but the functional implication of many of the associated genes remains unclear. N6-Methyladenosine (m6A) is the most abundant internal modification in mRNA. m6A methylation regulates many aspects of mRNA metabolism, playing important roles in the development of several pathologies. Interestingly, SNPs located near or within m6A motifs have been proposed as possible contributors to disease pathogenesis. -

Mir-17-92 Fine-Tunes MYC Expression and Function to Ensure
ARTICLE Received 31 Mar 2015 | Accepted 22 Sep 2015 | Published 10 Nov 2015 DOI: 10.1038/ncomms9725 OPEN miR-17-92 fine-tunes MYC expression and function to ensure optimal B cell lymphoma growth Marija Mihailovich1, Michael Bremang1, Valeria Spadotto1, Daniele Musiani1, Elena Vitale1, Gabriele Varano2,w, Federico Zambelli3, Francesco M. Mancuso1,w, David A. Cairns1,w, Giulio Pavesi3, Stefano Casola2 & Tiziana Bonaldi1 The synergism between c-MYC and miR-17-19b, a truncated version of the miR-17-92 cluster, is well-documented during tumor initiation. However, little is known about miR-17-19b function in established cancers. Here we investigate the role of miR-17-19b in c-MYC-driven lymphomas by integrating SILAC-based quantitative proteomics, transcriptomics and 30 untranslated region (UTR) analysis upon miR-17-19b overexpression. We identify over one hundred miR-17-19b targets, of which 40% are co-regulated by c-MYC. Downregulation of a new miR-17/20 target, checkpoint kinase 2 (Chek2), increases the recruitment of HuR to c- MYC transcripts, resulting in the inhibition of c-MYC translation and thus interfering with in vivo tumor growth. Hence, in established lymphomas, miR-17-19b fine-tunes c-MYC activity through a tight control of its function and expression, ultimately ensuring cancer cell homeostasis. Our data highlight the plasticity of miRNA function, reflecting changes in the mRNA landscape and 30 UTR shortening at different stages of tumorigenesis. 1 Department of Experimental Oncology, European Institute of Oncology, Via Adamello 16, Milan 20139, Italy. 2 Units of Genetics of B cells and lymphomas, IFOM, FIRC Institute of Molecular Oncology Foundation, Milan 20139, Italy. -

Trna GENES AFFECT MULTIPLE ASPECTS of LOCAL CHROMOSOME BEHAVIOR
tRNA GENES AFFECT MULTIPLE ASPECTS OF LOCAL CHROMOSOME BEHAVIOR by Matthew J Pratt-Hyatt A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in The University of Michigan 2008 Doctoral Committee: Professor David R. Engelke, Chair Professor Robert S. Fuller Associate Professor Mats E. Ljungman Associate Professor Thomas E. Wilson Assistant Professor Daniel A. Bochar © Matthew J. Pratt-Hyatt All rights reserved 2008 . To my loving family Who’ve helped me so much ii Acknowledgements I would first like to thank my mentor, David Engelke. His help has been imperative to the development of my scientific reasoning, writing, and public speaking. He has been incredibly supportive at each phase of my graduate career here at the University of Michigan. Second, I would like to thank the past and present members of the Engelke lab. I would especially like to thank Paul Good, May Tsoi, Glenn Wozniak, Kevin Kapadia, and Becky Haeusler for all of their help. For this dissertation I would like to thank the following people for their assistance. For Chapter II, I would like to thank Kevin Kapadia and Paul Good for technical assistance. I would also like to thank David Engelke and Tom Wilson for help with critical thinking and writing. For Chapter III, I would like to thank David Engelke and Rebecca Haeusler for their major writing contribution. I would also like to acknowledge that Rebecca Haeusler did the majority of the work that led to figures 1-3 in that chapter. For Chapter IV, I would like to thank Anita Hopper, David Engelke and Rebecca Haeusler for their writing contributions. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Revostmm Vol 10-4-2018 Ingles Maquetaciûn 1
108 ORIGINALS / Rev Osteoporos Metab Miner. 2018;10(4):108-18 Roca-Ayats N1, Falcó-Mascaró M1, García-Giralt N2, Cozar M1, Abril JF3, Quesada-Gómez JM4, Prieto-Alhambra D5,6, Nogués X2, Mellibovsky L2, Díez-Pérez A2, Grinberg D1, Balcells S1 1 Departamento de Genética, Microbiología y Estadística - Facultad de Biología - Universidad de Barcelona - Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) - Instituto de Salud Carlos III (ISCIII) - Instituto de Biomedicina de la Universidad de Barcelona (IBUB) - Instituto de Investigación Sant Joan de Déu (IRSJD) - Barcelona (España) 2 Unidad de Investigación en Fisiopatología Ósea y Articular (URFOA); Instituto Hospital del Mar de Investigaciones Médicas (IMIM) - Parque de Salud Mar - Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES); Instituto de Salud Carlos III (ISCIII) - Barcelona (España) 3 Departamento de Genética, Microbiología y Estadística; Facultad de Biología; Universidad de Barcelona - Instituto de Biomedicina de la Universidad de Barcelona (IBUB) - Barcelona (España) 4 Unidad de Metabolismo Mineral; Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC); Hospital Universitario Reina Sofía - Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES); Instituto de Salud Carlos III (ISCIII) - Córdoba (España) 5 Grupo de Investigación en Enfermedades Prevalentes del Aparato Locomotor (GREMPAL) - Instituto de Investigación en Atención Primaria (IDIAP) Jordi Gol - Centro de Investigación