1 Identification and Functional Characterisation of Genetic Variants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

9, 2015 Glasgow, Scotland, United Kingdom Abstracts
Volume 23 Supplement 1 June 2015 www.nature.com/ejhg European Human Genetics Conference 2015 June 6 - 9, 2015 Glasgow, Scotland, United Kingdom Abstracts EJHG_OFC.indd 1 4/1/2006 10:58:05 AM ABSTRACTS European Human Genetics Conference joint with the British Society of Genetics Medicine June 6 - 9, 2015 Glasgow, Scotland, United Kingdom Abstracts ESHG 2015 | GLASGOW, SCOTLAND, UK | WWW.ESHG.ORG 1 ABSTRACTS Committees – Board - Organisation European Society of Human Genetics ESHG Office Executive Board 2014-2015 Scientific Programme Committee European Society President Chair of Human Genetics Helena Kääriäinen, FI Brunhilde Wirth, DE Andrea Robinson Vice-President Members Karin Knob Han Brunner, NL Tara Clancy, UK c/o Vienna Medical Academy Martina Cornel, NL Alser Strasse 4 President-Elect Yanick Crow, FR 1090 Vienna Feliciano Ramos, ES Paul de Bakker, NL Austria Secretary-General Helene Dollfus, FR T: 0043 1 405 13 83 20 or 35 Gunnar Houge, NO David FitzPatrick, UK F: 0043 1 407 82 74 Maurizio Genuardi, IT E: [email protected] Deputy-Secretary-General Daniel Grinberg, ES www.eshg.org Karin Writzl, SI Gunnar Houge, NO Treasurer Erik Iwarsson, SE Andrew Read, UK Xavier Jeunemaitre, FR Mark Longmuir, UK Executive Officer Jose C. Machado, PT Jerome del Picchia, AT Dominic McMullan, UK Giovanni Neri, IT William Newman, UK Minna Nyström, FI Pia Ostergaard, UK Francesc Palau, ES Anita Rauch, CH Samuli Ripatti, FI Peter N. Robinson, DE Kristel van Steen, BE Joris Veltman, NL Joris Vermeesch, BE Emma Woodward, UK Karin Writzl, SI Board Members Liaison Members Yasemin Alanay, TR Stan Lyonnet, FR Martina Cornel, NL Martijn Breuning, NL Julie McGaughran, AU Ulf Kristoffersson, SE Pascal Borry, BE Bela Melegh, HU Thomas Liehr, DE Nina Canki-Klain, HR Will Newman, UK Milan Macek Jr., CZ Ana Carrió, ES Markus Nöthen, DE Tayfun Ozcelik, TR Isabella Ceccherini, IT Markus Perola, FI Milena Paneque, PT Angus John Clarke, UK Dijana Plaseska-Karanfilska, MK Hans Scheffer, NL Koen Devriendt, BE Trine E. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Placenta-Derived Exosomes Continuously Increase in Maternal
Sarker et al. Journal of Translational Medicine 2014, 12:204 http://www.translational-medicine.com/content/12/1/204 RESEARCH Open Access Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy Suchismita Sarker1, Katherin Scholz-Romero1, Alejandra Perez2, Sebastian E Illanes1,2,3, Murray D Mitchell1, Gregory E Rice1,2 and Carlos Salomon1,2* Abstract Background: Human placenta releases specific nanovesicles (i.e. exosomes) into the maternal circulation during pregnancy, however, the presence of placenta-derived exosomes in maternal blood during early pregnancy remains to be established. The aim of this study was to characterise gestational age related changes in the concentration of placenta-derived exosomes during the first trimester of pregnancy (i.e. from 6 to 12 weeks) in plasma from women with normal pregnancies. Methods: A time-series experimental design was used to establish pregnancy-associated changes in maternal plasma exosome concentrations during the first trimester. A series of plasma were collected from normal healthy women (10 patients) at 6, 7, 8, 9, 10, 11 and 12 weeks of gestation (n = 70). We measured the stability of these vesicles by quantifying and observing their protein and miRNA contents after the freeze/thawing processes. Exosomes were isolated by differential and buoyant density centrifugation using a sucrose continuous gradient and characterised by their size distribution and morphology using the nanoparticles tracking analysis (NTA; Nanosight™) and electron microscopy (EM), respectively. The total number of exosomes and placenta-derived exosomes were determined by quantifying the immunoreactive exosomal marker, CD63 and a placenta-specific marker (Placental Alkaline Phosphatase PLAP). -

The Cavefish Genome Reveals Candidate Genes for Eye Loss
ARTICLE Received 31 Jul 2014 | Accepted 17 Sep 2014 | Published 20 Oct 2014 DOI: 10.1038/ncomms6307 OPEN The cavefish genome reveals candidate genes for eye loss Suzanne E. McGaugh1,w, Joshua B. Gross2, Bronwen Aken3,4, Maryline Blin5, Richard Borowsky6, Domitille Chalopin7,He´le`ne Hinaux5, William R. Jeffery8, Alex Keene9,LiMa8, Patrick Minx1, Daniel Murphy3,4, Kelly E. O’Quin10, Sylvie Re´taux5, Nicolas Rohner11, Steve M.J. Searle3, Bethany A. Stahl2, Cliff Tabin11, Jean-Nicolas Volff7, Masato Yoshizawa9 & Wesley C. Warren1 Natural populations subjected to strong environmental selection pressures offer a window into the genetic underpinnings of evolutionary change. Cavefish populations, Astyanax mexicanus (Teleostei: Characiphysi), exhibit repeated, independent evolution for a variety of traits including eye degeneration, pigment loss, increased size and number of taste buds and mechanosensory organs, and shifts in many behavioural traits. Surface and cave forms are interfertile making this system amenable to genetic interrogation; however, lack of a reference genome has hampered efforts to identify genes responsible for changes in cave forms of A. mexicanus. Here we present the first de novo genome assembly for Astyanax mexicanus cavefish, contrast repeat elements to other teleost genomes, identify candidate genes underlying quantitative trait loci (QTL), and assay these candidate genes for potential functional and expression differences. We expect the cavefish genome to advance under- standing of the evolutionary process, as well as, analogous human disease including retinal dysfunction. 1 The Genome Institute, Washington University, Campus Box 8501, St Louis, Missouri 63108, USA. 2 Department of Biological Sciences, University of Cincinnati, 711B Rieveschl Hall, 312 College Drive, Cincinnati, Ohio 45221, USA. -

(12) United States Patent (10) Patent No.: US 9,057,109 B2 Chang (45) Date of Patent: Jun
US009057109B2 (12) United States Patent (10) Patent No.: US 9,057,109 B2 Chang (45) Date of Patent: Jun. 16, 2015 (54) DIAGNOSIS OF MELANOMA AND SOLAR 5,989,815 A 11/1999 Skolnicket al. LENTIGO BY NUCLEC ACID ANALYSIS 6,054,277 A 4/2000 Furcht et al. 6,056,859 A 5/2000 Ramsey et al. 6,129,983 A 10/2000 Schumann et al. (71) Applicant: DermTech International, La Jolla, CA 6,203,987 B1 3/2001 Friend et al. (US) 6,312.909 B1 1 1/2001 Shyjan 6,355.439 B1 3/2002 Chung et al. (72) Inventor: Sherman H. Chang, San Diego, CA 6.410,019 B1 6/2002 DeSimone et al. 6,410,240 B1 6/2002 Hodge et al. (US) 6,551,799 B2 4/2003 Gurney et al. 6,720,145 B2 4/2004 Rheins et al. (73) Assignee: DERMTECH INTERNATIONAL, La 6,726,971 B1 4/2004 Wong Jolla, CA (US) 6,891,022 B1 5/2005 Steward et al. 6,949,338 B2 9, 2005 Rheins et al. (*) Notice: Subject to any disclaimer, the term of this 7,183,057 B2 2/2007 Benson 7,247.426 B2 7/2007 Yakhini et al. patent is extended or adjusted under 35 7,267,951 B2 9, 2007 Alani et al. U.S.C. 154(b) by 0 days. 7,297,480 B2 11/2007 Vogt 7,615,349 B2 11/2009 Riker et al. (21) Appl. No.: 14/199,900 7,662,558 B2 2/2010 Liew 7.919,246 B2 * 4/2011 Lai et al. -

Trajectories of DNA Methylation Associated with Clozapine Exposure and Clinical Outcomes
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Trajectories of DNA Methylation Associated with Clozapine Exposure and Clinical Outcomes Gillespie, Amy Louise Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 List of Tables TRAJECTORIES OF DNA METHYLATION ASSOCIATED WITH CLOZAPINE EXPOSURE AND CLINICAL OUTCOMES Amy Gillespie INSTITUTE OF PSYCHIATRY PSYCHOLOGY AND NEUROSCIENCE, KING'S COLLEGE - 1 - List of Tables Contents List of Tables .................................................................................................................................... -

Whole Transcriptome Analysis on Blue Light-Induced Eye Damage
Blue light-induced eye damage ·Basic Research· Whole transcriptome analysis on blue light-induced eye damage Xin-Li Ouyang1, Bo-Yu Chen2, Yong-Fang Xie1, Yi-De Wu1, Shao-Jia Guo1, Xiao-Yun Dong1, Guo-Hui Wang1 1Key Laboratory of Biological Medicines in Universities of signaling pathways in the eye. Shandong Province, Weifang Medical University, Weifang ● KEYWORDS: blue light; eye; whole transcriptome 261053, Shandong Province, China sequencing; gene expression; signaling pathways; mice 2 Shijiazhuang Aier Eye Hospital, Bethune International Peace DOI:10.18240/ijo.2020.08.06 Hospital of PLA, Shijiazhuang 050082, Hebei Province, China Co-first authors: Xin-Li Ouyang, Bo-Yu Chen, and Yong- Citation: Ouyang XL, Chen BY, Xie YF, Wu YD, Guo SJ, Dong XY, Fang Xie Wang GH. Whole transcriptome analysis on blue light-induced eye Correspondence to: Guo-Hui Wang and Xiao-Yun Dong. damage. Int J Ophthalmol 2020;13(8):1210-1222 Key Laboratory of Biological Medicines in Universities of Shandong Province, Weifang Medical University, Weifang INTRODUCTION 261053, Shandong Province, China. wangguohui1983@163. xposure to blue light induces various biochemical and com; [email protected] Ephysiological changes in the eyes. In previous studies, Received: 2020-03-30 Accepted: 2020-05-07 blue light has been found to have high photochemical energy, and long exposure times to this high energy light often lead Abstract to eye diseases such as age-related maculopathy, dry eye and ● AIM: To analyze abnormal gene expressions of mice eyes [1-5] cataracts , and it will cause a worsening of visual fatigue exposed to blue light using RNA-seq and analyze the related and nearsightedness[6-7]. -
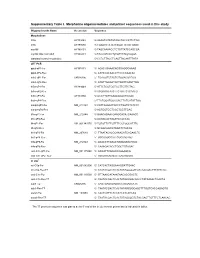
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Genomic Signatures of Drift and Selection Driven by Predation and Human Pressure in an Insular Lizard Marta Bassitta 1*, Richard P
www.nature.com/scientificreports OPEN Genomic signatures of drift and selection driven by predation and human pressure in an insular lizard Marta Bassitta 1*, Richard P. Brown 2, Ana Pérez‑Cembranos 3, Valentín Pérez‑Mellado 3, José A. Castro 1, Antònia Picornell 1 & Cori Ramon 1 Genomic divergence was studied in 10 small insular populations of the endangered Balearic Islands lizard (Podarcis lilfordi) using double digest restriction‑site associated DNA sequencing. The objectives were to establish levels of divergence among populations, investigate the impact of population size on genetic variability and to evaluate the role of diferent environmental factors on local adaptation. Analyses of 72,846 SNPs supported a highly diferentiated genetic structure, being the populations with the lowest population size (Porros, Foradada and Esclatasang islets) the most divergent, indicative of greater genetic drift. Outlier tests identifed ~ 2% of loci as candidates for selection. Genomic divergence‑Enviroment Association analyses were performed using redundancy analyses based on SNPs putatively under selection, detecting predation and human pressure as the environmental variables with the greatest explanatory power. Geographical distributions of populations and environmental factors appear to be fundamental drivers of divergence. These results support the combined role of genetic drift and divergent selection in shaping the genetic structure of these endemic island lizard populations. Insular populations are naturally isolated systems that harbour high levels of biodiversity and endemism1. Teir characteristic isolation leads to a reduction in gene fow and generates population divergence and speciation2. High levels of genetic structuring also result from frequent physical events combined with the impact of rapid fxation rates in ofen small populations subject to genetic drif and selection3,4. -

Ctbp Represses Adult Β-Like Globin Expression in Haematopoiesis
CtBP represses adult -like globin expression in haematopoiesis Jinfen Jasmine Yik A thesis submitted for the degree of Doctor of Philosophy (Biotechnology) School of Biotechnology and Biomolecular Sciences University of New South Wales October 2017 ORIGINALITY STATEMENT i Table of Contents Table of Contents ORIGINALITY STATEMENT ......................................................................................................... i Acknowledgements ................................................................................................................... v Publications arising from this candidature ................................................................................ vi Abstract .................................................................................................................................. vii List of abbreviations ............................................................................................................... viii 1 Chapter 1: Introduction .................................................................................................... 1 1.1 Mammalian gene regulation ..................................................................................... 1 1.2 Gene regulatory elements......................................................................................... 1 1.3 Transcription factors ................................................................................................. 2 1.4 Haematopoiesis ....................................................................................................... -

Identification and Characterization of Downstream Effector Protein(S) Regulated by P53 and Prb Miranda B
Marshall University Marshall Digital Scholar Theses, Dissertations and Capstones 2014 Identification and characterization of downstream effector protein(s) regulated by p53 and pRb Miranda B. Carper [email protected] Follow this and additional works at: http://mds.marshall.edu/etd Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, and the Medical Cell Biology Commons Recommended Citation Carper, Miranda B., "Identification and characterization of downstream effector protein(s) regulated by p53 and pRb" (2014). Theses, Dissertations and Capstones. Paper 897. This Dissertation is brought to you for free and open access by Marshall Digital Scholar. It has been accepted for inclusion in Theses, Dissertations and Capstones by an authorized administrator of Marshall Digital Scholar. For more information, please contact [email protected]. IDENTIFICATION AND CHARACTERIZATION OF DOWNSTREAM EFFECTOR PROTEIN(S) REGULATED BY p53 AND pRb A dissertation submitted to the Graduate College of Marshall University In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences By Miranda B. Carper Approved by Pier Paolo Claudio, MD, PhD, Committee Chairperson Richard Niles, PhD W. Elaine Hardman, PhD Jagan Valluri, PhD Piyali Dasgupta, PhD Emine Koc, PhD Marshall University December, 2014 DEDICATION My thesis is dedicated to my father, Millard. He is the foundation in my life in which I gather my strength. Without his love, support, and encouraging words none of this would have been possible. ii ACKNOWLEDGMENTS “The mediocre teacher tells. The good teacher explains. The superior teacher demonstrates. The great teacher inspires.” ― William Arthur Ward I would like to first acknowledge my advisor and mentor Dr. -

Title Over Expression of NANOS3 and DAZL in Human Embryonic Stem Cells Author(S) Panula, Sarita; Reda, Ahmed; Stukenborg, Jan-Be
Over Expression of NANOS3 and DAZL in Human Embryonic Title Stem Cells Panula, Sarita; Reda, Ahmed; Stukenborg, Jan-Bernd; Ramathal, Cyril; Sukhwani, Meena; Albalushi, Halima; Author(s) Edsgärd, Daniel; Nakamura, Michiko; Söder, Olle; Orwig, Kyle E.; Yamanaka, Shinya; Reijo Pera, Renee A.; Hovatta, Outi Citation PLOS ONE (2016), 11(10) Issue Date 2016-10-21 URL http://hdl.handle.net/2433/218666 © 2016 Panula et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, Right which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Type Journal Article Textversion publisher Kyoto University RESEARCH ARTICLE Over Expression of NANOS3 and DAZL in Human Embryonic Stem Cells Sarita Panula1, Ahmed Reda2, Jan-Bernd Stukenborg2, Cyril Ramathal3, Meena Sukhwani4, Halima Albalushi2,5, Daniel EdsgaÈrd6, Michiko Nakamura7, Olle SoÈ der2, Kyle E. Orwig4, Shinya Yamanaka7,8, Renee A. Reijo Pera3,9, Outi Hovatta1* 1 Division of Obstetrics and Gynecology, Department of Clinical Sciences, Intervention and Technology, Karolinska Institutet and Karolinska University Hospital, Huddinge, SE-141 86, Stockholm, Sweden, 2 Pediatric Endocrinology Unit, Department of Women's and Children's Health, Karolinska Institutet and Karolinska University Hospital, SE-171 76, Stockholm, Sweden, 3 Department of Genetics and Department of Obstetrics and Gynecology, Institute for Stem Cell Biology and Regenerative Medicine, Center for a11111 Reproductive