Luigi D'ascenzo, Ph.D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Open-Sourced Bioinformatic Pipeline for the Processing of Next-Generation Sequencing Derived Nucleotide Reads
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.20.050369; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. An open-sourced bioinformatic pipeline for the processing of Next-Generation Sequencing derived nucleotide reads: Identification and authentication of ancient metagenomic DNA Thomas C. Collin1, *, Konstantina Drosou2, 3, Jeremiah Daniel O’Riordan4, Tengiz Meshveliani5, Ron Pinhasi6, and Robin N. M. Feeney1 1School of Medicine, University College Dublin, Ireland 2Division of Cell Matrix Biology Regenerative Medicine, University of Manchester, United Kingdom 3Manchester Institute of Biotechnology, School of Earth and Environmental Sciences, University of Manchester, United Kingdom [email protected] 5Institute of Paleobiology and Paleoanthropology, National Museum of Georgia, Tbilisi, Georgia 6Department of Evolutionary Anthropology, University of Vienna, Austria *Corresponding Author Abstract The emerging field of ancient metagenomics adds to these Bioinformatic pipelines optimised for the processing and as- processing complexities with the need for additional steps sessment of metagenomic ancient DNA (aDNA) are needed in the separation and authentication of ancient sequences from modern sequences. Currently, there are few pipelines for studies that do not make use of high yielding DNA cap- available for the analysis of ancient metagenomic DNA ture techniques. These bioinformatic pipelines are tradition- 1 4 ally optimised for broad aDNA purposes, are contingent on (aDNA) ≠ The limited number of bioinformatic pipelines selection biases and are associated with high costs. -
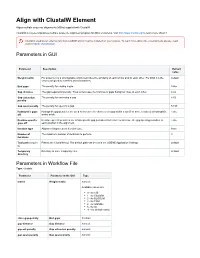
Align with Clustalw Element
Align with ClustalW Element Aligns multiple sequence alignments (MSAs) supplied with ClustalW. ClustalW is a general purpose multiple sequence alignment program for DNA or proteins. Visit http://www.clustal.org/ to learn more about it. Clustal is used as an external tool from UGENE and it must be installed on your system. To learn more about the external tools, please, read main UGENE User Manual. Parameters in GUI Parameter Description Default value Weight matrix For proteins it is a scoring table which describes the similarity of each amino acid to each other. For DNA it is the default scores assigned to matches and mismatches. End gaps The penalty for closing a gap. False Gap distance The gap separation penalty. Tries to decrease the chances of gaps being too close to each other. 4.42 Gap extension The penalty for extending a gap. 8.52 penalty Gap open penalty The penalty for opening a gap. 53.90 Hydrophilic gaps Hydrophilic gap penalties are used to increase the chances of a gap within a run (5 or more residues) of hydrophilic False off amino acids. Residue-specific Residue-specific penalties are amino specific gap penalties that reduce or increase the gap opening penalties at False gaps off each position in the alignment. Iteration type Alignment improvement iteration type. None Number of The maximum number of iterations to perform. 3 iterations Tool path (require Path to the ClustalW tool. The default path can be set in the UGENE Application Settings. default d) Temporary Directory to store temporary files. default directory Parameters -

Real Time Collaboration on Multiple Sequence Alignments Jeffrey Yunes Submitted in Partial Satisfaction of the Require
ShareMSA: real time collaboration on multiple sequence alignments Jeffrey Yunes Submitted in partial satisfaction of the requirements for the degree of m in the GRADUATE DIVISION UNIVERSITY OF CALIFORNIA, SAN FRANCISCO ii Acknowledgements I am very thankful for the guidance and support of my thesis advisor and chair, Patricia C. Babbitt. I thank my thesis committee members Hao Li and Ian Holmes. I am grateful to Elaine Meng for feedback on user experience and Eric Pettersen for help with Chimera plugins. This work was funded by NIH GM60595 to Patricia C. Babbitt. iii Table of Contents Chapter1.................................................. 1 Abstract................................................. 1 Introduction............................................... 1 Features................................................. 1 ExamplesofUse ............................................ 2 Conclusion ............................................... 4 References . 5 iv List of Figures Figure 1 Screenshot from the mobile version of the site . 3 v Abstract A Multiple Sequence Alignment (MSA) is a widely used data structure in bioinformatics for applications ranging from evolutionary analyses to protein engineering. ShareMSA is a website that allows users to visualize, share, and collaborate on MSAs. ShareMSA makes MSAs more accessible by allowing collaborators one-click access to the MSA and annotations without the need to install software. In addition, when those with requisite permissions change the annotations or legend, all viewers of the MSA see the changes in real time. A Chimera plugin is available via the ShareMSA website, allowing Chimera users to publish MSAs from within this sequence-structure visualization software. Availability and Implementation ShareMSA is freely available at https://www.sharemsa.com, and does not require registration. The front end is implemented in HTML and JavaScript and runs on all modern browsers that support WebSockets. -

A User-Friendly Sequence Editor for Mac OS X
Fourment and Holmes BMC Res Notes (2016) 9:106 DOI 10.1186/s13104-016-1927-4 BMC Research Notes TECHNICAL NOTE Open Access Seqotron: a user‑friendly sequence editor for Mac OS X Mathieu Fourment1,2* and Edward C. Holmes2 Abstract Background: Accurate multiple sequence alignment is central to bioinformatics and molecular evolutionary analy- ses. Although sophisticated sequence alignment programs are available, manual adjustments are often required to improve alignment quality. Unfortunately, few programs offer a simple and intuitive way to edit sequence alignments. Results: We present Seqotron, a sequence editor that reads and writes files in a wide variety of sequence formats. Sequences can be easily aligned and manually edited using the mouse and keyboard. The program also allows the user to estimate both phylogenetic trees and distance matrices. Conclusions: Seqotron will benefit researchers who need to manipulate and align complex sequence data. Seqotron is a Mac OS X compatible open source project and is available from Github https://github.com/4ment/seqotron/. Keywords: Sequence editor, Alignment, Phylogenetics Background file formats. Alignments can be generated automatically State-of-the-art methods of multiple sequence alignment using the MUSCLE [1] and MAFFT [2] packages and the such as MUSCLE [1] and MAFFT [2] are usually used to quality of the alignment can be visually inspected and automatically generate alignments. Unfortunately, these manually corrected using simple mouse-based and key- methods can be inaccurate when the input sequences are board-based operations. In addition, Seqotron allows the highly dissimilar or when sequencing errors have been computation of distance matrices and the inference of incorporated. -

Download E-Book (PDF)
OPEN ACCESS African Journal of Biotechnology 17 April 2019 ISSN 1684-5315 DOI: 10.5897/AJB www.academicjournals.org About AJB The African Journal of Biotechnology (AJB) is a peer reviewed journal which commenced publication in 2002. AJB publishes articles from all areas of biotechnology including medical and pharmaceutical biotechnology, molecular diagnostics, applied biochemistry, industrial microbiology, molecular biology, bioinformatics, genomics and proteomics, transcriptomics and genome editing, food and agricultural technologies, and metabolic engineering. Manuscripts on economic and ethical issues relating to biotechnology research are also considered. Indexing CAB Abstracts, CABI’s Global Health Database, Chemical Abstracts (CAS Source Index) Dimensions Database, Google Scholar, Matrix of Information for The Analysis of Journals (MIAR), Microsoft Academic, Research Gate Open Access Policy Open Access is a publication model that enables the dissemination of research articles to the global community without restriction through the internet. All articles published under open access can be accessed by anyone with internet connection. The African Journals of Biotechnology is an Open Access journal. Abstracts and full texts of all articles published in this journal are freely accessible to everyone immediately after publication without any form of restriction. Article License All articles published by African Journal of Biotechnology are licensed under the Creative Commons Attribution 4.0 International License. This permits anyone to -

The Plastid and Mitochondrial Genomes of Eucalyptus Grandis Desre Pinard1,2, Alexander A
Pinard et al. BMC Genomics (2019) 20:132 https://doi.org/10.1186/s12864-019-5444-4 RESEARCHARTICLE Open Access The plastid and mitochondrial genomes of Eucalyptus grandis Desre Pinard1,2, Alexander A. Myburg1,2 and Eshchar Mizrachi1,2* Abstract Background: Land plant organellar genomes have significant impact on metabolism and adaptation, and as such, accurate assembly and annotation of plant organellar genomes is an important tool in understanding the evolutionary history and interactions between these genomes. Intracellular DNA transfer is ongoing between the nuclear and organellar genomes, and can lead to significant genomic variation between, and within, species that impacts downstream analysis of genomes and transcriptomes. Results: In order to facilitate further studies of cytonuclear interactions in Eucalyptus, we report an updated annotation of the E. grandis plastid genome, and the second sequenced and annotated mitochondrial genome of the Myrtales, that of E. grandis. The 478,813 bp mitochondrial genome shows the conserved protein coding regions and gene order rearrangements typical of land plants. There have been widespread insertions of organellar DNA into the E. grandis nuclear genome, which span 141 annotated nuclear genes. Further, we identify predicted editing sites to allow for the discrimination of RNA-sequencing reads between nuclear and organellar gene copies, finding that nuclear copies of organellar genes are not expressed in E. grandis. Conclusions: The implications of organellar DNA transfer to the nucleus are often ignored, despite the insight they can give into the ongoing evolution of plant genomes, and the problems they can cause in many applications of genomics. Future comparisons of the transcription and regulation of organellar genes between Eucalyptus genotypes may provide insight to the cytonuclear interactions that impact economically important traits in this widely grown lignocellulosic crop species. -

A Review of the Smith-Waterman GPU Landscape
A Review of the Smith-Waterman GPU Landscape Richard Barnes Electrical Engineering and Computer Sciences University of California at Berkeley Technical Report No. UCB/EECS-2020-152 http://www2.eecs.berkeley.edu/Pubs/TechRpts/2020/EECS-2020-152.html August 13, 2020 Copyright © 2020, by the author(s). All rights reserved. Permission to make digital or hard copies of all or part of this work for personal or classroom use is granted without fee provided that copies are not made or distributed for profit or commercial advantage and that copies bear this notice and the full citation on the first page. To copy otherwise, to republish, to post on servers or to redistribute to lists, requires prior specific permission. Acknowledgement Richard Barnes was supported by the Department of Energy's Computational Science Graduate Fellowship (Grant DE-FG02-97ER25308) and, through the Berkeley Institute for Data Science, by the Gordon and Betty Moore Foundation (Grant GBMF3834) and by the Alfred P. Sloan Foundation (Grant 2013-10-27). Tests were performed on the Titan and Summitdev supercomputers managed by Oak Ridge National Laboratory's Leadership Computing Facility; the Cori supercomputer managed by the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231; and XSEDE's Comet supercomputer, which is supported by the NSF (Grant ACI- 1053575) and was available to RB through the NSF Graduate Research Fellowship. AReviewoftheSmith-WatermanGPULandscape Richard Barnes Research Project Submitted to the Department of Electrical Engineering and Computer Sci- ences, University of California at Berkeley, in partial satisfaction of the re- quirements for the degree of Master of Science, Plan II. -

Expertdiscovery and UGENE Integrated System for Intelligent Analysis of Regulatory Regions of Genes
In Silico Biology 11 (2011/2012) 97–108 97 DOI 10.3233/ISB-2012-0448 IOS Press ExpertDiscovery and UGENE integrated system for intelligent analysis of regulatory regions of genes Y.Y. Vaskina,*, I.V. Khomichevab, E.V. Ignatievac and E.E. Vityaevb aNovosibirsk State University, Department of Information Technology, Novosibirsk, Russia bInstitute of Mathematics, SD RAS, Novosibirsk, Russia cInstitute of Cytology and Genetics, SD RAS, Novosibirsk, Russia Abstract. The task of automatic extraction of the hierarchical structure of eukaryotic gene regulatory regions is in the junction of the fields of biology, mathematics and information technologies. A solution of the problem involves understanding of sophis- ticated mechanisms of eukaryotic gene regulation and applying advanced data mining technologies. In the paper the integrated system, implementing a powerful relation mining of biological data method, is discussed. The system allows taking into account prior information about the gene regulatory regions that is known by the biologist, performing the analysis on each hierarchical level, searching for a solution from a simple hypothesis to a complex one. The integration of ExpertDiscovery system into UGENE toolkit provides a convenient environment for conducting complex research and automating the work of a biologist. For demonstration, the system has been applied for recognition of SF1, SREBP, HNF4 vertebrate binding sites and for the analysis the human gene regulatory regions that promote liver-specific transcription. Keywords: Complex signal, hierarchical analysis, recognition, gene regulatory regions, bioinformatics 1. Introduction RNA molecule (a primary transcript) with essentially the same sequence as the gene. Given the complexity Analysis of gene regulatory regions and searching for of multicellular eukaryotes, transcriptional regulation structural and functional patterns are actual problems of in these organisms needs to be very complex [30]. -
Shared Bioinformatics Databases Within the Unipro UGENE Platform
Journal of Integrative Bioinformatics, 12(1):257, 2015 http://journal.imbio.de Shared Bioinformatics Databases within the Unipro UGENE Platform Ivan V. Protsyuk 1,2*, German A. Grekhov 1, Alexey V. Tiunov 1 and Mikhail Yu. Fursov 1 1Unipro Center for Information Technologies, 6/1 Lavrentieva Ave., 630090 Novosibirsk, Russia 2Department of Physics, Novosibirsk State University, 2 Pirogova St., 630090 Novosibirsk, Russia Summary Unipro UGENE is an open-source bioinformatics toolkit that integrates popular tools along with original instruments for molecular biologists within a unified user interface. Nowadays, most bioinformatics desktop applications, including UGENE, make use of a local data model while processing different types of data. Such an approach causes an (http://creativecommons.org/licenses/by-nc-nd/3.0/). inconvenience for scientists working cooperatively and relying on the same data. This refers to the need of making multiple copies of certain files for every workplace and License maintaining synchronization between them in case of modifications. Therefore, we focused on delivering a collaborative work into the UGENE user experience. Currently, several UGENE installations can be connected to a designated shared database and users can interact with it simultaneously. Such databases can be created by UGENE users and be used at their discretion. Objects of each data type, supported by UGENE such as sequences, annotations, multiple alignments, etc., can now be easily imported from or exported to a remote storage. One of the main advantages of this system, compared to existing ones, is the almost simultaneous access of client applications to shared data regardless of their volume. Moreover, the system is capable of storing millions of objects. -
• Sequence Database Searching • Phylogenetic Trees • Protein Structure
Prof. Bystroff talks about BIOINFORMATICS hi • Sequence database searching • Phylogenetic Trees • Protein Structure 1 AAAGAGATTCTGCTAGCGGTCGG A G A G A T G C T G C A G C G A G T C G G C C AAAGAGATTCTGCTAGCGGTCGG A G A G A T G C T G C A G C G A G T C G G C C AAAGAGATTCTGCTAGCGGTCGG A G A G A T G C T G C A G C G A G T C G G C C Protein sequence alignment uses a "substitution matrix". Sequence 1 Sequence2 5 Find the best pathway through the substitution scores, and you have an alignment 6 "dynamic programming" algorithm. BLAST searches millions of sequences GenBank contains over 162 million sequences!! The score for each should be the optimal alignment score. Even if we can do 1 per millisecond, it would take 45 hours to do one search. BLAST usually finishes in under a minute. How does BLAST do it so fast? BLAST precalculates all triplet hits in the database. ... PGQ PGR PGS ... My sequence has PGT PGV PGW this triplet BLAST saves a lookup table (called PGY PAQ PCQ an INDEX) for all of PGQ the near identity PDQ PEQ PFQ ... triplet location in the ... whole database. BLAST uses an expansion table to allow for near perfect matches This is all done when BLAST is set up, before any searches are carried out. BLAST finds diagonal arrangements of triplet hits triplet hits in one database Hits are joined by extension protein BLAST scores only the best hits (saves time) cutoff Re-scoring. -
Unipro UGENE Online User Manual
Unipro UGENE Online User Manual About Unipro About UGENE Key Features User Interface High Performance Computing Cooperation Download and Installation System Requirements UGENE Packages Installation on Windows Installation on Mac OS X Installation on Linux Native Installation on Ubuntu Native Installation on Fedora Basic Functions UGENE Terminology UGENE Window Components Welcome Page Project View Task View Log View Notifications Main Menu Overview Creating New Project Creating Document Opening Document Opening for the First Time Advanced Dialog Options Opening Document Present in Project Opening Several Documents Opening Containing Folder Exporting Documents Locked Documents Using Objects and Object Views Exporting Objects Exporting Sequences to Sequence Format Exporting Sequences as Alignment Exporting Alignment to Sequence Format Exporting Nucleic Alignment to Amino Translation Export Sequences Associated with Annotation Using Bookmarks Exporting Project Search in Project Options Panel Adding and Removing Plugins Searching NCBI Genbank Fetching Data from Remote Database UGENE Application Settings General Resources Network File Format Directories Logging Alignment Color Scheme External Tools Settings Genome Aligner Workflow Designer Settings OpenCL Sequence View Sequence View Components Global Actions Sequence Toolbars Sequence Overview Sequence Zoom View Sequence Details View Information about Sequence Manipulating Sequence Going To Position Toggling Views Exporting Sequence Image Zooming Sequence Creating New Ruler Selecting Amino Translation -

Ugene, a Newly Identified Protein That Is Commonly Overexpressed in Cancer and Binds Uracil DNA Glycosylase
Research Article Ugene, a Newly Identified Protein That Is Commonly Overexpressed in Cancer and Binds Uracil DNA Glycosylase Chunguang Guo,1,4 Xiaodong Zhang,3,4 Stephen P. Fink,2,4 Petra Platzer,2,4 Keith Wilson,7 James K.V. Willson,8 Zhenghe Wang,3,4 and Sanford D. Markowitz1,2,4,5,6 Departments of 1Molecular Biology and Microbiology, 2Medicine, and 3Genetics; 4Case Comprehensive Cancer Center, Case Western Reserve University; 5Case Medical Center; 6Howard Hughes Medical Institute, Cleveland, Ohio; 7PDL BioPHARMA, Redwood City, California; and 8Harold C. Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas Abstract complex with uracil DNA glycosylase 2 (UNG2), a base excision Expression microarrays identified a novel transcript, desig- repair (BER) enzyme. nated as Ugene, whose expression is absent in normal colon and colon adenomas, but that is commonly induced in Materials and Methods malignant colon cancers. These findings were validated by Cell lines and tissues. VACO cell lines were established and maintained real-time PCR and Northern blot analysis in an independent as previously described (7). DLD1 and SW480 cell lines were obtained from panel of colon cancer cases. In addition, Ugene expression was the American Type Culture Collection. Normal colons, primary colon found to be elevated in many other common cancer types, cancers, and liver metastasis tissues were obtained from the archives of including breast, lung, uterus, and ovary. Immunofluores- University Hospitals of Cleveland under an institutional review board– cence of V5-tagged Ugene revealed it to have a nuclear approved protocol. Total RNA and genomic DNA were prepared as described (8).