A Review of the Smith-Waterman GPU Landscape
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioinformatics Study of Lectins: New Classification and Prediction In
Bioinformatics study of lectins : new classification and prediction in genomes François Bonnardel To cite this version: François Bonnardel. Bioinformatics study of lectins : new classification and prediction in genomes. Structural Biology [q-bio.BM]. Université Grenoble Alpes [2020-..]; Université de Genève, 2021. En- glish. NNT : 2021GRALV010. tel-03331649 HAL Id: tel-03331649 https://tel.archives-ouvertes.fr/tel-03331649 Submitted on 2 Sep 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE L’UNIVERSITE GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et l’Université de Genève Spécialités: Chimie Biologie Arrêté ministériel : le 6 janvier 2005 – 25 mai 2016 Présentée par François Bonnardel Thèse dirigée par la Dr. Anne Imberty codirigée par la Dr/Prof. Frédérique Lisacek préparée au sein du laboratoire CERMAV, CNRS et du Computer Science Department, UNIGE et de l’équipe PIG, SIB Dans les Écoles Doctorales EDCSV et UNIGE Etude bioinformatique des lectines: nouvelle classification et prédiction dans les génomes Thèse soutenue publiquement le 8 Février 2021, devant le jury composé de : Dr. Alexandre de Brevern UMR S1134, Inserm, Université Paris Diderot, Paris, France, Rapporteur Dr. -

A SARS-Cov-2 Sequence Submission Tool for the European Nucleotide
Databases and ontologies Downloaded from https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab421/6294398 by guest on 25 June 2021 A SARS-CoV-2 sequence submission tool for the European Nucleotide Archive Miguel Roncoroni 1,2,∗, Bert Droesbeke 1,2, Ignacio Eguinoa 1,2, Kim De Ruyck 1,2, Flora D’Anna 1,2, Dilmurat Yusuf 3, Björn Grüning 3, Rolf Backofen 3 and Frederik Coppens 1,2 1Department of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Ghent, Belgium, 1VIB Center for Plant Systems Biology, 9052 Ghent, Belgium and 2University of Freiburg, Department of Computer Science, Freiburg im Breisgau, Baden-Württemberg, Germany ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Summary: Many aspects of the global response to the COVID-19 pandemic are enabled by the fast and open publication of SARS-CoV-2 genetic sequence data. The European Nucleotide Archive (ENA) is the European recommended open repository for genetic sequences. In this work, we present a tool for submitting raw sequencing reads of SARS-CoV-2 to ENA. The tool features a single-step submission process, a graphical user interface, tabular-formatted metadata and the possibility to remove human reads prior to submission. A Galaxy wrap of the tool allows users with little or no bioinformatic knowledge to do bulk sequencing read submissions. The tool is also packed in a Docker container to ease deployment. Availability: CLI ENA upload tool is available at github.com/usegalaxy- eu/ena-upload-cli (DOI 10.5281/zenodo.4537621); Galaxy ENA upload tool at toolshed.g2.bx.psu.edu/view/iuc/ena_upload/382518f24d6d and https://github.com/galaxyproject/tools- iuc/tree/master/tools/ena_upload (development) and; ENA upload Galaxy container at github.com/ELIXIR- Belgium/ena-upload-container (DOI 10.5281/zenodo.4730785) Contact: [email protected] 1 Introduction Nucleotide Archive (ENA). -

An Open-Sourced Bioinformatic Pipeline for the Processing of Next-Generation Sequencing Derived Nucleotide Reads
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.20.050369; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. An open-sourced bioinformatic pipeline for the processing of Next-Generation Sequencing derived nucleotide reads: Identification and authentication of ancient metagenomic DNA Thomas C. Collin1, *, Konstantina Drosou2, 3, Jeremiah Daniel O’Riordan4, Tengiz Meshveliani5, Ron Pinhasi6, and Robin N. M. Feeney1 1School of Medicine, University College Dublin, Ireland 2Division of Cell Matrix Biology Regenerative Medicine, University of Manchester, United Kingdom 3Manchester Institute of Biotechnology, School of Earth and Environmental Sciences, University of Manchester, United Kingdom [email protected] 5Institute of Paleobiology and Paleoanthropology, National Museum of Georgia, Tbilisi, Georgia 6Department of Evolutionary Anthropology, University of Vienna, Austria *Corresponding Author Abstract The emerging field of ancient metagenomics adds to these Bioinformatic pipelines optimised for the processing and as- processing complexities with the need for additional steps sessment of metagenomic ancient DNA (aDNA) are needed in the separation and authentication of ancient sequences from modern sequences. Currently, there are few pipelines for studies that do not make use of high yielding DNA cap- available for the analysis of ancient metagenomic DNA ture techniques. These bioinformatic pipelines are tradition- 1 4 ally optimised for broad aDNA purposes, are contingent on (aDNA) ≠ The limited number of bioinformatic pipelines selection biases and are associated with high costs. -

Six-Fold Speed-Up of Smith-Waterman Sequence Database Searches Using Parallel Processing on Common Microprocessors
Six-fold speed-up of Smith-Waterman sequence database searches using parallel processing on common microprocessors Running head: Six-fold speed-up of Smith-Waterman searches Torbjørn Rognes* and Erling Seeberg Institute of Medical Microbiology, University of Oslo, The National Hospital, NO-0027 Oslo, Norway Abstract Motivation: Sequence database searching is among the most important and challenging tasks in bioinformatics. The ultimate choice of sequence search algorithm is that of Smith- Waterman. However, because of the computationally demanding nature of this method, heuristic programs or special-purpose hardware alternatives have been developed. Increased speed has been obtained at the cost of reduced sensitivity or very expensive hardware. Results: A fast implementation of the Smith-Waterman sequence alignment algorithm using SIMD (Single-Instruction, Multiple-Data) technology is presented. This implementation is based on the MMX (MultiMedia eXtensions) and SSE (Streaming SIMD Extensions) technology that is embedded in Intel’s latest microprocessors. Similar technology exists also in other modern microprocessors. Six-fold speed-up relative to the fastest previously known Smith-Waterman implementation on the same hardware was achieved by an optimised 8-way parallel processing approach. A speed of more than 150 million cell updates per second was obtained on a single Intel Pentium III 500MHz microprocessor. This is probably the fastest implementation of this algorithm on a single general-purpose microprocessor described to date. Availability: Online searches with the software are available at http://dna.uio.no/search/ Contact: [email protected] Published in Bioinformatics (2000) 16 (8), 699-706. Copyright © (2000) Oxford University Press. *) To whom correspondence should be addressed. -

Bioinformatics Courses Lecture 3: (Local) Alignment and Homology
C E N Bioinformatics courses T R E Principles of Bioinformatics (BSc) & F B O I Fundamentals of Bioinformatics R O I I N N (MSc) T F E O G R R M A A T T I I Lecture 3: (local) alignment and V C E S homology searching V U Centre for Integrative Bioinformatics VU (IBIVU) Faculty of Exact Sciences / Faculty of Earth and Life Sciences 1 http://ibi.vu.nl, [email protected], 87649 (Heringa), Room P1.28 Divergent evolution sequence -> structure -> function • Common ancestor (CA) CA • Sequences change over time • Protein structures typically remain the same (robust against Sequence 1 ≠ Sequence 2 multiple mutations) • Therefore, function normally is Structure 1 = Structure 2 preserved within orthologous families Function 1 = Function 2 “Structure more conserved than sequence” 2 Reconstructing divergent evolution Ancestral sequence: ABCD ACCD (B C) ABD (C ø) mutation deletion ACCD or ACCD Pairwise Alignment AB─D A─BD 3 Reconstructing divergent evolution Ancestral sequence: ABCD ACCD (B C) ABD (C ø) mutation deletion ACCD or ACCD Pairwise Alignment AB─D A─BD true alignment 4 Pairwise alignment examples A protein sequence alignment MSTGAVLIY--TSILIKECHAMPAGNE----- ---GGILLFHRTHELIKESHAMANDEGGSNNS * * * **** *** A DNA sequence alignment attcgttggcaaatcgcccctatccggccttaa att---tggcggatcg-cctctacgggcc---- *** **** **** ** ****** 5 Evolution and three-dimensional protein structure information Multiple alignment Protein structure What do we see if we colour code the space-filling (CPK) protein model? • E.g., red for conserved alignment positions to blue for variable (unconserved) positions. 6 Evolution and three-dimensional protein structure information Isocitrate dehydrogenase: The distance from the active site (in yellow) determines the rate of evolution (red = fast evolution, blue = slow evolution) Dean, A. -
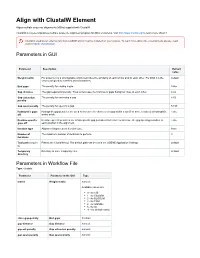
Align with Clustalw Element
Align with ClustalW Element Aligns multiple sequence alignments (MSAs) supplied with ClustalW. ClustalW is a general purpose multiple sequence alignment program for DNA or proteins. Visit http://www.clustal.org/ to learn more about it. Clustal is used as an external tool from UGENE and it must be installed on your system. To learn more about the external tools, please, read main UGENE User Manual. Parameters in GUI Parameter Description Default value Weight matrix For proteins it is a scoring table which describes the similarity of each amino acid to each other. For DNA it is the default scores assigned to matches and mismatches. End gaps The penalty for closing a gap. False Gap distance The gap separation penalty. Tries to decrease the chances of gaps being too close to each other. 4.42 Gap extension The penalty for extending a gap. 8.52 penalty Gap open penalty The penalty for opening a gap. 53.90 Hydrophilic gaps Hydrophilic gap penalties are used to increase the chances of a gap within a run (5 or more residues) of hydrophilic False off amino acids. Residue-specific Residue-specific penalties are amino specific gap penalties that reduce or increase the gap opening penalties at False gaps off each position in the alignment. Iteration type Alignment improvement iteration type. None Number of The maximum number of iterations to perform. 3 iterations Tool path (require Path to the ClustalW tool. The default path can be set in the UGENE Application Settings. default d) Temporary Directory to store temporary files. default directory Parameters -

A Survey on Data Compression Methods for Biological Sequences
Article A Survey on Data Compression Methods for Biological Sequences Morteza Hosseini *, Diogo Pratas and Armando J. Pinho Institute of Electronics and Informatics Engineering of Aveiro/Department of Electronics, Telecommunications and Informatics (IEETA/DETI), University of Aveiro, 3810-193 Aveiro, Portugal; [email protected] (D.P.); [email protected] (A.J.P.) * Correspondence: [email protected]; Tel.: +351-234-370520; Fax: +351-234-370545 Academic Editor: Khalid Sayood Received: 27 June 2016; Accepted: 29 September 2016; Published: 14 October 2016 Abstract: The ever increasing growth of the production of high-throughput sequencing data poses a serious challenge to the storage, processing and transmission of these data. As frequently stated, it is a data deluge. Compression is essential to address this challenge—it reduces storage space and processing costs, along with speeding up data transmission. In this paper, we provide a comprehensive survey of existing compression approaches, that are specialized for biological data, including protein and DNA sequences. Also, we devote an important part of the paper to the approaches proposed for the compression of different file formats, such as FASTA, as well as FASTQ and SAM/BAM, which contain quality scores and metadata, in addition to the biological sequences. Then, we present a comparison of the performance of several methods, in terms of compression ratio, memory usage and compression/decompression time. Finally, we present some suggestions for future research on biological data compression. Keywords: protein sequence; DNA sequence; reference-free compression; reference-based compression; FASTA; Multi-FASTA; FASTQ; SAM; BAM 1. Introduction With the development of high-throughput sequencing (HTS) technologies, huge volumes of sequencing data are produced everyday, fast and cheaply, that can lead to upgrading the scientific knowledge of genome sequence information as well as facilitating the diagnosis and therapy. -

Biohackathon Series in 2013 and 2014
F1000Research 2019, 8:1677 Last updated: 02 AUG 2021 OPINION ARTICLE BioHackathon series in 2013 and 2014: improvements of semantic interoperability in life science data and services [version 1; peer review: 2 approved with reservations] Toshiaki Katayama 1, Shuichi Kawashima1, Gos Micklem 2, Shin Kawano 1, Jin-Dong Kim1, Simon Kocbek1, Shinobu Okamoto1, Yue Wang1, Hongyan Wu3, Atsuko Yamaguchi 1, Yasunori Yamamoto1, Erick Antezana 4, Kiyoko F. Aoki-Kinoshita5, Kazuharu Arakawa6, Masaki Banno7, Joachim Baran8, Jerven T. Bolleman 9, Raoul J. P. Bonnal10, Hidemasa Bono 1, Jesualdo T. Fernández-Breis11, Robert Buels12, Matthew P. Campbell13, Hirokazu Chiba14, Peter J. A. Cock15, Kevin B. Cohen16, Michel Dumontier17, Takatomo Fujisawa18, Toyofumi Fujiwara1, Leyla Garcia 19, Pascale Gaudet9, Emi Hattori20, Robert Hoehndorf21, Kotone Itaya6, Maori Ito22, Daniel Jamieson23, Simon Jupp19, Nick Juty19, Alex Kalderimis2, Fumihiro Kato 24, Hideya Kawaji25, Takeshi Kawashima18, Akira R. Kinjo26, Yusuke Komiyama27, Masaaki Kotera28, Tatsuya Kushida 29, James Malone30, Masaaki Matsubara 31, Satoshi Mizuno32, Sayaka Mizutani 28, Hiroshi Mori33, Yuki Moriya1, Katsuhiko Murakami34, Takeru Nakazato1, Hiroyo Nishide14, Yosuke Nishimura 28, Soichi Ogishima32, Tazro Ohta1, Shujiro Okuda35, Hiromasa Ono1, Yasset Perez-Riverol 19, Daisuke Shinmachi5, Andrea Splendiani36, Francesco Strozzi37, Shinya Suzuki 28, Junichi Takehara28, Mark Thompson38, Toshiaki Tokimatsu39, Ikuo Uchiyama 14, Karin Verspoor 40, Mark D. Wilkinson 41, Sarala Wimalaratne19, Issaku Yamada -

Database and Bioinformatic Analysis of BCL-2 Family Proteins and BH3-Only Proteins Abdel Aouacheria, Vincent Navratil, Christophe Combet
Database and Bioinformatic Analysis of BCL-2 Family Proteins and BH3-Only Proteins Abdel Aouacheria, Vincent Navratil, Christophe Combet To cite this version: Abdel Aouacheria, Vincent Navratil, Christophe Combet. Database and Bioinformatic Analysis of BCL-2 Family Proteins and BH3-Only Proteins. Gavathiotis E. BCL-2 Family Proteins. Methods in Molecular Biology, 1877, Springer Nature, pp.23-43, 2019, Methods in Molecular Biology, 978-1-4939- 8860-0. 10.1007/978-1-4939-8861-7_2. hal-02347884 HAL Id: hal-02347884 https://hal.archives-ouvertes.fr/hal-02347884 Submitted on 5 Nov 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Copyright Database and bioinformatic analysis of BCL-2 family proteins and BH3-only proteins Abdel Aouacheria 1,*, Vincent Navratil 2 and Christophe Combet 3 1 ISEM, Institut des Sciences de l’Evolution de Montpellier, Université de Montpellier, UMR 5554, CNRS, IRD, EPHE, Place Eugène Bataillon, 34095 Montpellier, France 2 PRABI, Rhône Alpes Bioinformatics Center, UCBL, Lyon1, Université de Lyon, Lyon, France. 3 Centre de Recherche en Cancérologie de Lyon, UMR Inserm U1052, CNRS 5286, Université Claude Bernard Lyon 1, Centre Léon Bérard, Lyon, France. -

Protein Database
UniProt Unit-2 Paper- Bioinformatics (DSE-1) B.Sc. (H) Microbiology V Sem UniProt It provide the scientific community with a comprehensive, high-quality and freely accessible resource of protein sequence and functional information UniProt - Universal protein resource Swiss-Prot & TrEMBL PIR-PSD •Merger of these three databases, since 2002 •Funded mainly by NIH (US) to be the highest quality, most thoroughly annotated protein sequence database •UniProt is a collaboration between the European Bioinformatics Institute (EMBL- EBI), the Swiss Institute of Bioinformatics (SIB) and the Protein Information Resource (PIR). •Collaboration through different tasks such as database curation, software development and support. Source of protein sequence data • Protein sequencing is rare • Most protein sequence Large-scale sequencing Individual Patent derived from nucleotide data projects scientists Offices Nucleotide sequencing Submit Protein sequencing Submit Nucleotide Derive Protein sequence protein sequence sequence database database More than 95 % of the protein sequences provided by UniProtKB are derived from the translation of the coding sequences (CDS) Protein sequence is mainly derived data submit DNA sequence ACGCTCGTACGCATCGTCACTACTAGCTACGACGACGACACGCTACTACTCGACGATTCT may not have direct Predicted Predicted transcribe Predicted evidence start splice sites stop Derived mRNA sequence AUGCGUAGUGAUGAAUGCUGCUGUGCGAUGAGCUGC translate Derived protein sequence MRSNECCCAMSC UniProt • The Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data. • The UniProt databases are: 1. UniProt Knowledgebase (UniProtKB), 2. UniProt Reference Clusters (UniRef), 3. UniProt Archive (UniParc). UniProtKB UniProtKB • Reviewed (Swiss-Prot) - Manually annotated Records with information extracted from literature and curator-evaluated computational analysis and and scientific conclusions. • Is a high quality non-redundant protein sequence database • Unreviewed (TrEMBL) - Computationally analyzed Records that await full manual annotation. -

Sequence Database Searching
Sequence Database Searching 2007-01-14 Gao, Ge Center for Bioinformatics, Peking University Sequence Alignment: A Quick Review Object: see how close two sequences are Usages: To infer functions. To determine the common area of some sequences To infer protein structure, if the structure of one of the sequences is known. To “guess” whether they are descended from a common ancestor (and construct evolutionary trees). © Copyright 2006 Center for Bioinformatics, Peking University Sequence Alignment: in Biology The purpose of a sequence alignment is to line up all residues in the sequence that were derived from the same residue position in the ancestral gene or protein in any number of sequences A B A B © Copyright 2006g aCpe n=te irn fsoer rBtiooinf oorrm dateicles,t iPoenking University Sequence Alignment: Scoring GAATC GAAT-C -GAAT-C CATAC C-ATAC C-A-TAC GAATC- GAAT-C GA-ATC CA-TAC CA-TAC CATA-C Scoring function: measure the quality of a candidate alignment. scoring matrix, gap penalty © Copyright 2006 Center for Bioinformatics, Peking University Sequence Alignment : Global vs. Local Global alignments: align residues in whole sequence, are most useful when the sequences are similar and of roughly equal size. Algorithm: Needleman-Wunsch Local alignments: align residues in regions are more useful for diverged sequences Algorithm: Smith-Waterman With sufficiently similar sequences, there is no difference between local and global alignments. © Copyright 2006 Center for Bioinformatics, Peking University Sequence Database Searching Rather than do the alignment pair-wise, it’s necessary to run database searching in a high-throughput style. Identify similarities between novel query sequences whose structures and functions are unknown and uncharacterized sequences in (public) databases whose structures and functions have been elucidated. -

Accessing Molecular Biology Information * Genome Browsers
Accessing molecular biology information * Genome browsers - UCSC * NCBI * Galaxy Flow of genetic information and databases Exon 1 Exon 2 Exon 3 Exon 4 intron intron intron DNA transcription 5' 3' RNA Genbank/ EMBL splicing 5' UTR 3' UTR polyA mature coding sequence mRNA translation protein GenPept / SwissProt folding / UniProt Protein Data Bank (PDB) Databases, cont. Redundancy at GenBank => RefSeq Many sequences are represented more than once in GenBank 2003 RefSeq collection : curated secondary database non-redundant selected organisms •Genome DNA (assemblies) •Transcripts (RNA) •Protein http://www.ncbi.nlm.nih.gov/books/bv.fcg i?rid=handbook RefSeq vs GenBank GenBank RefSeq Not curated Curated Author submits NCBI creates from existing data Only author can revise NCBI reivses as new data emerge Multiple records from same loci Single records for each molecule common of major organisms Records can contradict each other No limit to species included Limited to model organisms Akin to primary literature Akin to review articles Archives with nucleotide sequences • Genbank/EMBL • Sequence read archive (NCBI) • GEO (Gene expression omnibus, NCBI) • 1000 Genomes Project • The Cancer Genome Atlas • The Cancer Genome Project • Species-specific databases like –FlyBase – WormBase – Saccharomyces Genome Database Genome sequencing using a shotgun approach Sequenced eukaryotic genomes Sequencing going wild ... BGI : "capacity to sequence the equivalent of 1,600 complete human genomes each day" "BGI and BGI Americas aim to build a library of digital