UGENE – a Practical Approach for Complex Computational Analysis in Molecular Biology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Effective Computational Method Incorporating Multiple Secondary
Old Dominion University ODU Digital Commons Computer Science Faculty Publications Computer Science 2017 An Effective Computational Method Incorporating Multiple Secondary Structure Predictions in Topology Determination for Cryo-EM Images Abhishek Biswas Old Dominion University Desh Ranjan Old Dominion University Mohammad Zubair Old Dominion University Stephanie Zeil Old Dominion University Kamal Al Nasr See next page for additional authors Follow this and additional works at: https://digitalcommons.odu.edu/computerscience_fac_pubs Part of the Biochemistry Commons, Computer Sciences Commons, Molecular Biology Commons, and the Statistics and Probability Commons Repository Citation Biswas, Abhishek; Ranjan, Desh; Zubair, Mohammad; Zeil, Stephanie; Al Nasr, Kamal; and He, Jing, "An Effective Computational Method Incorporating Multiple Secondary Structure Predictions in Topology Determination for Cryo-EM Images" (2017). Computer Science Faculty Publications. 81. https://digitalcommons.odu.edu/computerscience_fac_pubs/81 Original Publication Citation Biswas, A., Ranjan, D., Zubair, M., Zeil, S., Al Nasr, K., & He, J. (2017). An effective computational method incorporating multiple secondary structure predictions in topology determination for cryo-em images. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 14(3), 578-586. doi:10.1109/tcbb.2016.2543721 This Article is brought to you for free and open access by the Computer Science at ODU Digital Commons. It has been accepted for inclusion in Computer Science Faculty Publications by an authorized administrator of ODU Digital Commons. For more information, please contact [email protected]. Authors Abhishek Biswas, Desh Ranjan, Mohammad Zubair, Stephanie Zeil, Kamal Al Nasr, and Jing He This article is available at ODU Digital Commons: https://digitalcommons.odu.edu/computerscience_fac_pubs/81 HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author IEEE/ACM Manuscript Author Trans Comput Manuscript Author Biol Bioinform. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Increasing the Accuracy of Single Sequence Prediction Methods Using a Deep Semi-Supervised Learning Framework Lewis Moffat 1,∗ and David T
Structural Bioinformatics Downloaded from https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btab491/6313164 by guest on 07 July 2021 Increasing the Accuracy of Single Sequence Prediction Methods Using a Deep Semi-Supervised Learning Framework Lewis Moffat 1,∗ and David T. Jones 1,∗ 1Department of Computer Science, University College London, Gower Street, London WC1E 6BT, UK; and Francis Crick Institute, 1 Midland Road, London NW1 1AT, UK ∗To whom correspondence should be addressed. Associate Editor: XXXXXXX Received on XXXXX; revised on XXXXX; accepted on XXXXX Abstract Motivation: Over the past 50 years, our ability to model protein sequences with evolutionary information has progressed in leaps and bounds. However, even with the latest deep learning methods, the modelling of a critically important class of proteins, single orphan sequences, remains unsolved. Results: By taking a bioinformatics approach to semi-supervised machine learning, we develop Profile Augmentation of Single Sequences (PASS), a simple but powerful framework for building accurate single- sequence methods. To demonstrate the effectiveness of PASS we apply it to the mature field of secondary structure prediction. In doing so we develop S4PRED, the successor to the open-source PSIPRED-Single method, which achieves an unprecedented Q3 score of 75.3% on the standard CB513 test. PASS provides a blueprint for the development of a new generation of predictive methods, advancing our ability to model individual protein sequences. Availability: The S4PRED model is available as open source software on the PSIPRED GitHub repository (https://github.com/psipred/s4pred), along with documentation. It will also be provided as a part of the PSIPRED web service (http://bioinf.cs.ucl.ac.uk/psipred/) Contact: [email protected], [email protected] Supplementary information: Supplementary data are available at Bioinformatics online. -

An Open-Sourced Bioinformatic Pipeline for the Processing of Next-Generation Sequencing Derived Nucleotide Reads
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.20.050369; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. An open-sourced bioinformatic pipeline for the processing of Next-Generation Sequencing derived nucleotide reads: Identification and authentication of ancient metagenomic DNA Thomas C. Collin1, *, Konstantina Drosou2, 3, Jeremiah Daniel O’Riordan4, Tengiz Meshveliani5, Ron Pinhasi6, and Robin N. M. Feeney1 1School of Medicine, University College Dublin, Ireland 2Division of Cell Matrix Biology Regenerative Medicine, University of Manchester, United Kingdom 3Manchester Institute of Biotechnology, School of Earth and Environmental Sciences, University of Manchester, United Kingdom [email protected] 5Institute of Paleobiology and Paleoanthropology, National Museum of Georgia, Tbilisi, Georgia 6Department of Evolutionary Anthropology, University of Vienna, Austria *Corresponding Author Abstract The emerging field of ancient metagenomics adds to these Bioinformatic pipelines optimised for the processing and as- processing complexities with the need for additional steps sessment of metagenomic ancient DNA (aDNA) are needed in the separation and authentication of ancient sequences from modern sequences. Currently, there are few pipelines for studies that do not make use of high yielding DNA cap- available for the analysis of ancient metagenomic DNA ture techniques. These bioinformatic pipelines are tradition- 1 4 ally optimised for broad aDNA purposes, are contingent on (aDNA) ≠ The limited number of bioinformatic pipelines selection biases and are associated with high costs. -

Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian
G C A T T A C G G C A T genes Article Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment Edmond Wonkam-Tingang 1, Isabelle Schrauwen 2 , Kevin K. Esoh 1 , Thashi Bharadwaj 2, Liz M. Nouel-Saied 2 , Anushree Acharya 2, Abdul Nasir 3 , Samuel M. Adadey 1,4 , Shaheen Mowla 5 , Suzanne M. Leal 2 and Ambroise Wonkam 1,* 1 Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa; [email protected] (E.W.-T.); [email protected] (K.K.E.); [email protected] (S.M.A.) 2 Center for Statistical Genetics, Sergievsky Center, Taub Institute for Alzheimer’s Disease and the Aging Brain, and the Department of Neurology, Columbia University Medical Centre, New York, NY 10032, USA; [email protected] (I.S.); [email protected] (T.B.); [email protected] (L.M.N.-S.); [email protected] (A.A.); [email protected] (S.M.L.) 3 Synthetic Protein Engineering Lab (SPEL), Department of Molecular Science and Technology, Ajou University, Suwon 443-749, Korea; [email protected] 4 West African Centre for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana, Accra LG 54, Ghana 5 Division of Haematology, Department of Pathology, Faculty of Health Sciences, University of Cape Town, Cape Town 7925, South Africa; [email protected] * Correspondence: [email protected]; Tel.: +27-21-4066-307 Received: 28 September 2020; Accepted: 20 October 2020; Published: 23 October 2020 Abstract: DNA samples from five members of a multiplex non-consanguineous Cameroonian family, segregating prelingual and progressive autosomal recessive non-syndromic sensorineural hearing impairment, underwent whole exome sequencing. -

Further Confirmation of the Association of SLC12A2 with Non-Syndromic
www.nature.com/jhg ARTICLE OPEN Further confirmation of the association of SLC12A2 with non-syndromic autosomal-dominant hearing impairment Samuel M. Adadey1,2,9, Isabelle Schrauwen3,9, Elvis Twumasi Aboagye1, Thashi Bharadwaj3, Kevin K. Esoh 2, Sulman Basit 4, 3 3 5 2 6 1 Anushree Acharya , Liz M. Nouel-Saied ,✉ Khurram Liaqat , Edmond✉ Wonkam-Tingang , Shaheen Mowla , Gordon A. Awandare , Wasim Ahmad 7, Suzanne M. Leal 3,8 and Ambroise Wonkam 2 © The Author(s) 2021 Congenital hearing impairment (HI) is genetically heterogeneous making its genetic diagnosis challenging. Investigation of novel HI genes and variants will enhance our understanding of the molecular mechanisms and to aid genetic diagnosis. We performed exome sequencing and analysis using DNA samples from affected members of two large families from Ghana and Pakistan, segregating autosomal-dominant (AD) non-syndromic HI (NSHI). Using in silico approaches, we modeled and evaluated the effect of the likely pathogenic variants on protein structure and function. We identified two likely pathogenic variants in SLC12A2, c.2935G>A:p.(E979K) and c.2939A>T:p.(E980V), which segregate with NSHI in a Ghanaian and Pakistani family, respectively. SLC12A2 encodes an ion transporter crucial in the homeostasis of the inner ear endolymph and has recently been reported to be implicated in syndromic and non-syndromic HI. Both variants were mapped to alternatively spliced exon 21 of the SLC12A2 gene. Exon 21 encodes for 17 residues in the cytoplasmatic tail of SLC12A2, is highly conserved between species, and preferentially expressed in cochlear tissues. A review of previous studies and our current data showed that out of ten families with either AD non-syndromic or syndromic HI, eight (80%) had variants within the 17 amino acid residue region of exon 21 (48 bp), suggesting that this alternate domain is critical to the transporter activity in the inner ear. -

Bayesian Model of Protein Primary Sequence for Secondary Structure Prediction
Bayesian Model of Protein Primary Sequence for Secondary Structure Prediction Qiwei Li1, David B. Dahl2*, Marina Vannucci1, Hyun Joo3, Jerry W. Tsai3 1 Department of Statistics, Rice University, Houston, Texas, United States of America, 2 Department of Statistics, Brigham Young University, Provo, Utah, United States of America, 3 Department of Chemistry, University of the Pacific, Stockton, California, United States of America Abstract Determining the primary structure (i.e., amino acid sequence) of a protein has become cheaper, faster, and more accurate. Higher order protein structure provides insight into a protein’s function in the cell. Understanding a protein’s secondary structure is a first step towards this goal. Therefore, a number of computational prediction methods have been developed to predict secondary structure from just the primary amino acid sequence. The most successful methods use machine learning approaches that are quite accurate, but do not directly incorporate structural information. As a step towards improving secondary structure reduction given the primary structure, we propose a Bayesian model based on the knob- socket model of protein packing in secondary structure. The method considers the packing influence of residues on the secondary structure determination, including those packed close in space but distant in sequence. By performing an assessment of our method on 2 test sets we show how incorporation of multiple sequence alignment data, similarly to PSIPRED, provides balance and improves the accuracy of the predictions. Software implementing the methods is provided as a web application and a stand-alone implementation. Citation: Li Q, Dahl DB, Vannucci M, Joo H, Tsai JW (2014) Bayesian Model of Protein Primary Sequence for Secondary Structure Prediction. -

The Structure and Dynamics of Bmr1 Protein from Brugia Malayi: in Silico Approaches
Int. J. Mol. Sci. 2014, 15, 11082-11099; doi:10.3390/ijms150611082 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches Bee Yin Khor, Gee Jun Tye, Theam Soon Lim, Rahmah Noordin and Yee Siew Choong * Institute for Research in Molecular Medicine, Universiti Sains Malaysia, Minden, Penang 11800, Malaysia; E-Mails: [email protected] (B.Y.K.); [email protected] (G.J.T.); [email protected] (T.S.L.); [email protected] (R.N.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +604-653-4837; Fax: +604-653-4803. Received: 28 January 2014; in revised form: 25 March 2014 / Accepted: 4 June 2014 / Published: 19 June 2014 Abstract: Brugia malayi is a filarial nematode, which causes lymphatic filariasis in humans. In 1995, the disease has been identified by the World Health Organization (WHO) as one of the second leading causes of permanent and long-term disability and thus it is targeted for elimination by year 2020. Therefore, accurate filariasis diagnosis is important for management and elimination programs. A recombinant antigen (BmR1) from the Bm17DIII gene product was used for antibody-based filariasis diagnosis in “Brugia Rapid”. However, the structure and dynamics of BmR1 protein is yet to be elucidated. Here we study the three dimensional structure and dynamics of BmR1 protein using comparative modeling, threading and ab initio protein structure prediction. The best predicted structure obtained via an ab initio method (Rosetta) was further refined and minimized. -

The PSIPRED Protein Structure Prediction Server Liam J
Vol. 16 no. 4 2000 BIOINFORMATICS APPLICATIONS NOTE Pages 404–405 The PSIPRED protein structure prediction server Liam J. McGuffin 1, 2, Kevin Bryson 1 and David T. Jones 1, 2 1Protein Bioinformatics Group, Department of Biological Sciences, University of Warwick, Coventry CV4 7AL, UK and 2Department of Biological Sciences, Brunel University, Uxbridge, UB8 3PH, UK Received on November 8, 1999; accepted on December 14, 1999 Abstract Prediction of secondary structure (PSIPRED) Summary: The PSIPRED protein structure prediction A new highly accurate secondary structure prediction server allows users to submit a protein sequence, perform method, PSIPRED incorporates two feed-forward neural a prediction of their choice and receive the results of the networks which perform an analysis on output obtained prediction both textually via e-mail and graphically via the from PSI-BLAST (Position Specific Iterated BLAST) web. The user may select one of three prediction methods (Altschul et al., 1997). to apply to their sequence: PSIPRED, a highly accurate Using a stringent cross validation procedure to evaluate secondary structure prediction method; MEMSAT 2, a performance, PSIPRED has been shown to be capable new version of a widely used transmembrane topology of achieving an average Q3 score of 76.5%. This is prediction method; or GenTHREADER, a sequence profile the highest level of accuracy published for any method based fold recognition method. to date. Predictions produced by PSIPRED were also Availability: Freely available to non-commercial users at submitted to the CASP3 (Third Critical Assessment http://globin.bio.warwick.ac.uk/psipred/ of Structure Prediction) server and assessed during the Contact: [email protected] CASP3 meeting, which took place in December 1998 at Asilomar. -

Performance of Secondary Structure Prediction Methods on Proteins
Performance of Secondary Structure Prediction Methods on Proteins PerformanceContaining of Structurally Secondary Ambivalent Structure Prediction Sequence Methods Fragments on Proteins Containing Structurally Ambivalent Sequence Fragments K. Mani Saravanan, Samuel Selvaraj Department of Bioinformatics, School of Life Sciences, Bharathidasan University, Tiruchirappalli 620024, Tamil Nadu, India Received 3 July 2012; revised 4 September 2012; accepted 23 September 2012 Published online 19 October 2012 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/bip.22178 # ABSTRACT: fragments. 2012 Wiley Periodicals, Inc. Biopolymers Several approaches for predicting secondary structures (Pept Sci) 100: 148-153, 2013. from sequences have been developed and reached a fair Keywords: octapeptides; secondary structure prediction; accuracy. One of the most rigorous tests for these segment overlap measure; structurally ambivalent frag- prediction methods is their ability to correctly predict ments; protein folding identical fragments of protein sequences adopting This article was originally published online as an accepted preprint. different secondary structures in unrelated proteins. In The ‘‘Published Online’’ date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers our previous work, we obtained 30 identical octapeptide editorial office at [email protected] sequence fragments adopting different backbone conformations. It is of interest to find whether the INTRODUCTION presence of structurally -

Biophysical Analysis of the N-Terminal Domain from The
This is a repository copy of Biophysical Analysis of the N-Terminal Domain from the Human Protein Phosphatase 1 Nuclear Targeting Subunit PNUTS Suggests an Extended Transcription Factor TFIIS-Like Fold. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/108536/ Version: Accepted Version Article: Zacharchenko, T, Barsukov, I, Rigden, DJ et al. (2 more authors) (2016) Biophysical Analysis of the N-Terminal Domain from the Human Protein Phosphatase 1 Nuclear Targeting Subunit PNUTS Suggests an Extended Transcription Factor TFIIS-Like Fold. Protein Journal, 35 (5). pp. 340-345. ISSN 1572-3887 https://doi.org/10.1007/s10930-016-9677-7 © 2016, Springer Science+Business Media New York. This is an author produced version of a paper published in Protein Journal. The final version is available at Springer via https://doi.org/10.1007/s10930-016-9677-7. Uploaded in accordance with the publisher's self-archiving policy. Reuse Unless indicated otherwise, fulltext items are protected by copyright with all rights reserved. The copyright exception in section 29 of the Copyright, Designs and Patents Act 1988 allows the making of a single copy solely for the purpose of non-commercial research or private study within the limits of fair dealing. The publisher or other rights-holder may allow further reproduction and re-use of this version - refer to the White Rose Research Online record for this item. Where records identify the publisher as the copyright holder, users can verify any specific terms of use on the publisher’s website. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. -
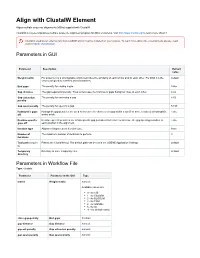
Align with Clustalw Element
Align with ClustalW Element Aligns multiple sequence alignments (MSAs) supplied with ClustalW. ClustalW is a general purpose multiple sequence alignment program for DNA or proteins. Visit http://www.clustal.org/ to learn more about it. Clustal is used as an external tool from UGENE and it must be installed on your system. To learn more about the external tools, please, read main UGENE User Manual. Parameters in GUI Parameter Description Default value Weight matrix For proteins it is a scoring table which describes the similarity of each amino acid to each other. For DNA it is the default scores assigned to matches and mismatches. End gaps The penalty for closing a gap. False Gap distance The gap separation penalty. Tries to decrease the chances of gaps being too close to each other. 4.42 Gap extension The penalty for extending a gap. 8.52 penalty Gap open penalty The penalty for opening a gap. 53.90 Hydrophilic gaps Hydrophilic gap penalties are used to increase the chances of a gap within a run (5 or more residues) of hydrophilic False off amino acids. Residue-specific Residue-specific penalties are amino specific gap penalties that reduce or increase the gap opening penalties at False gaps off each position in the alignment. Iteration type Alignment improvement iteration type. None Number of The maximum number of iterations to perform. 3 iterations Tool path (require Path to the ClustalW tool. The default path can be set in the UGENE Application Settings. default d) Temporary Directory to store temporary files. default directory Parameters