On Optimal Designs for Clinical Trials: an Updated Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Design of Experiments Application, Concepts, Examples: State of the Art
Periodicals of Engineering and Natural Scinces ISSN 2303-4521 Vol 5, No 3, December 2017, pp. 421‒439 Available online at: http://pen.ius.edu.ba Design of Experiments Application, Concepts, Examples: State of the Art Benjamin Durakovic Industrial Engineering, International University of Sarajevo Article Info ABSTRACT Article history: Design of Experiments (DOE) is statistical tool deployed in various types of th system, process and product design, development and optimization. It is Received Aug 3 , 2018 multipurpose tool that can be used in various situations such as design for th Revised Oct 20 , 2018 comparisons, variable screening, transfer function identification, optimization th Accepted Dec 1 , 2018 and robust design. This paper explores historical aspects of DOE and provides state of the art of its application, guides researchers how to conceptualize, plan and conduct experiments, and how to analyze and interpret data including Keyword: examples. Also, this paper reveals that in past 20 years application of DOE have Design of Experiments, been grooving rapidly in manufacturing as well as non-manufacturing full factorial design; industries. It was most popular tool in scientific areas of medicine, engineering, fractional factorial design; biochemistry, physics, computer science and counts about 50% of its product design; applications compared to all other scientific areas. quality improvement; Corresponding Author: Benjamin Durakovic International University of Sarajevo Hrasnicka cesta 15 7100 Sarajevo, Bosnia Email: [email protected] 1. Introduction Design of Experiments (DOE) mathematical methodology used for planning and conducting experiments as well as analyzing and interpreting data obtained from the experiments. It is a branch of applied statistics that is used for conducting scientific studies of a system, process or product in which input variables (Xs) were manipulated to investigate its effects on measured response variable (Y). -

Computing the Oja Median in R: the Package Ojanp
Computing the Oja Median in R: The Package OjaNP Daniel Fischer Karl Mosler Natural Resources Institute of Econometrics Institute Finland (Luke) and Statistics Finland University of Cologne and Germany School of Health Sciences University of Tampere Finland Jyrki M¨ott¨onen Klaus Nordhausen Department of Social Research Department of Mathematics University of Helsinki and Statistics Finland University of Turku Finland and School of Health Sciences University of Tampere Finland Oleksii Pokotylo Daniel Vogel Cologne Graduate School Institute of Complex Systems University of Cologne and Mathematical Biology Germany University of Aberdeen United Kingdom June 27, 2016 arXiv:1606.07620v1 [stat.CO] 24 Jun 2016 Abstract The Oja median is one of several extensions of the univariate median to the mul- tivariate case. It has many nice properties, but is computationally demanding. In this paper, we first review the properties of the Oja median and compare it to other multivariate medians. Afterwards we discuss four algorithms to compute the Oja median, which are implemented in our R-package OjaNP. Besides these 1 algorithms, the package contains also functions to compute Oja signs, Oja signed ranks, Oja ranks, and the related scatter concepts. To illustrate their use, the corresponding multivariate one- and C-sample location tests are implemented. Keywords Oja median, Oja signs, Oja signed ranks, Oja ranks, R,C,C++ 1 Introduction The univariate median is a popular location estimator. It is, however, not straightforward to generalize it to the multivariate case since no generalization known retains all properties of univariate estimator, and therefore different gen- eralizations emphasize different properties of the univariate median. -

Design of Experiments (DOE) Using JMP Charles E
PharmaSUG 2014 - Paper PO08 Design of Experiments (DOE) Using JMP Charles E. Shipp, Consider Consulting, Los Angeles, CA ABSTRACT JMP has provided some of the best design of experiment software for years. The JMP team continues the tradition of providing state-of-the-art DOE support. In addition to the full range of classical and modern design of experiment approaches, JMP provides a template for Custom Design for specific requirements. The other choices include: Screening Design; Response Surface Design; Choice Design; Accelerated Life Test Design; Nonlinear Design; Space Filling Design; Full Factorial Design; Taguchi Arrays; Mixture Design; and Augmented Design. Further, sample size and power plots are available. We give an introduction to these methods followed by a few examples with factors. BRIEF HISTORY AND BACKGROUND From early times, there has been a desire to improve processes with controlled experimentation. A famous early experiment cured scurvy with seamen taking citrus fruit. Lives were saved. Thomas Edison invented the light bulb with many experiments. These experiments depended on trial and error with no software to guide, measure, and refine experimentation. Japan led the way with improving manufacturing—Genichi Taguchi wanted to create repeatable and robust quality in manufacturing. He had been taught management and statistical methods by William Edwards Deming. Deming taught “Plan, Do, Check, and Act”: PLAN (design) the experiment; DO the experiment by performing the steps; CHECK the results by testing information; and ACT on the decisions based on those results. In America, other pioneers brought us “Total Quality” and “Six Sigma” with “Design of Experiments” being an advanced methodology. -
![Harmonizing Fully Optimal Designs with Classic Randomization in Fixed Trial Experiments Arxiv:1810.08389V1 [Stat.ME] 19 Oct 20](https://docslib.b-cdn.net/cover/7263/harmonizing-fully-optimal-designs-with-classic-randomization-in-fixed-trial-experiments-arxiv-1810-08389v1-stat-me-19-oct-20-167263.webp)
Harmonizing Fully Optimal Designs with Classic Randomization in Fixed Trial Experiments Arxiv:1810.08389V1 [Stat.ME] 19 Oct 20
Harmonizing Fully Optimal Designs with Classic Randomization in Fixed Trial Experiments Adam Kapelner, Department of Mathematics, Queens College, CUNY, Abba M. Krieger, Department of Statistics, The Wharton School of the University of Pennsylvania, Uri Shalit and David Azriel Faculty of Industrial Engineering and Management, The Technion October 22, 2018 Abstract There is a movement in design of experiments away from the classic randomization put forward by Fisher, Cochran and others to one based on optimization. In fixed-sample trials comparing two groups, measurements of subjects are known in advance and subjects can be divided optimally into two groups based on a criterion of homogeneity or \imbalance" between the two groups. These designs are far from random. This paper seeks to understand the benefits and the costs over classic randomization in the context of different performance criterions such as Efron's worst-case analysis. In the criterion that we motivate, randomization beats optimization. However, the optimal arXiv:1810.08389v1 [stat.ME] 19 Oct 2018 design is shown to lie between these two extremes. Much-needed further work will provide a procedure to find this optimal designs in different scenarios in practice. Until then, it is best to randomize. Keywords: randomization, experimental design, optimization, restricted randomization 1 1 Introduction In this short survey, we wish to investigate performance differences between completely random experimental designs and non-random designs that optimize for observed covariate imbalance. We demonstrate that depending on how we wish to evaluate our estimator, the optimal strategy will change. We motivate a performance criterion that when applied, does not crown either as the better choice, but a design that is a harmony between the two of them. -

Optimal Design of Experiments Session 4: Some Theory
Optimal design of experiments Session 4: Some theory Peter Goos 1 / 40 Optimal design theory Ï continuous or approximate optimal designs Ï implicitly assume an infinitely large number of observations are available Ï is mathematically convenient Ï exact or discrete designs Ï finite number of observations Ï fewer theoretical results 2 / 40 Continuous versus exact designs continuous Ï ½ ¾ x1 x2 ... xh Ï » Æ w1 w2 ... wh Ï x1,x2,...,xh: design points or support points w ,w ,...,w : weights (w 0, P w 1) Ï 1 2 h i ¸ i i Æ Ï h: number of different points exact Ï ½ ¾ x1 x2 ... xh Ï » Æ n1 n2 ... nh Ï n1,n2,...,nh: (integer) numbers of observations at x1,...,xn P n n Ï i i Æ Ï h: number of different points 3 / 40 Information matrix Ï all criteria to select a design are based on information matrix Ï model matrix 2 3 2 T 3 1 1 1 1 1 1 f (x1) ¡ ¡ Å Å Å T 61 1 1 1 1 17 6f (x2)7 6 Å ¡ ¡ Å Å 7 6 T 7 X 61 1 1 1 1 17 6f (x3)7 6 ¡ Å ¡ Å Å 7 6 7 Æ 6 . 7 Æ 6 . 7 4 . 5 4 . 5 T 100000 f (xn) " " " " "2 "2 I x1 x2 x1x2 x1 x2 4 / 40 Information matrix Ï (total) information matrix 1 1 Xn M XT X f(x )fT (x ) 2 2 i i Æ σ Æ σ i 1 Æ Ï per observation information matrix 1 T f(xi)f (xi) σ2 5 / 40 Information matrix industrial example 2 3 11 0 0 0 6 6 6 0 6 0 0 0 07 6 7 1 6 0 0 6 0 0 07 ¡XT X¢ 6 7 2 6 7 σ Æ 6 0 0 0 4 0 07 6 7 4 6 0 0 0 6 45 6 0 0 0 4 6 6 / 40 Information matrix Ï exact designs h X T M ni f(xi)f (xi) Æ i 1 Æ where h = number of different points ni = number of replications of point i Ï continuous designs h X T M wi f(xi)f (xi) Æ i 1 Æ 7 / 40 D-optimality criterion Ï seeks designs that minimize variance-covariance matrix of ¯ˆ ¯ 2 T 1¯ Ï . -
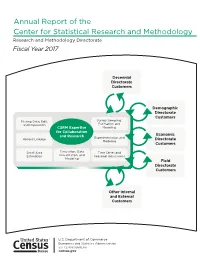
Annual Report of the Center for Statistical Research and Methodology Research and Methodology Directorate Fiscal Year 2017
Annual Report of the Center for Statistical Research and Methodology Research and Methodology Directorate Fiscal Year 2017 Decennial Directorate Customers Demographic Directorate Customers Missing Data, Edit, Survey Sampling: and Imputation Estimation and CSRM Expertise Modeling for Collaboration Economic and Research Experimentation and Record Linkage Directorate Modeling Customers Small Area Simulation, Data Time Series and Estimation Visualization, and Seasonal Adjustment Modeling Field Directorate Customers Other Internal and External Customers ince August 1, 1933— S “… As the major figures from the American Statistical Association (ASA), Social Science Research Council, and new Roosevelt academic advisors discussed the statistical needs of the nation in the spring of 1933, it became clear that the new programs—in particular the National Recovery Administration—would require substantial amounts of data and coordination among statistical programs. Thus in June of 1933, the ASA and the Social Science Research Council officially created the Committee on Government Statistics and Information Services (COGSIS) to serve the statistical needs of the Agriculture, Commerce, Labor, and Interior departments … COGSIS set … goals in the field of federal statistics … (It) wanted new statistical programs—for example, to measure unemployment and address the needs of the unemployed … (It) wanted a coordinating agency to oversee all statistical programs, and (it) wanted to see statistical research and experimentation organized within the federal government … In August 1933 Stuart A. Rice, President of the ASA and acting chair of COGSIS, … (became) assistant director of the (Census) Bureau. Joseph Hill (who had been at the Census Bureau since 1900 and who provided the concepts and early theory for what is now the methodology for apportioning the seats in the U.S. -

Concepts Underlying the Design of Experiments
Concepts Underlying the Design of Experiments March 2000 University of Reading Statistical Services Centre Biometrics Advisory and Support Service to DFID Contents 1. Introduction 3 2. Specifying the objectives 5 3. Selection of treatments 6 3.1 Terminology 6 3.2 Control treatments 7 3.3 Factorial treatment structure 7 4. Choosing the sites 9 5. Replication and levels of variation 10 6. Choosing the blocks 12 7. Size and shape of plots in field experiments 13 8. Allocating treatment to units 14 9. Taking measurements 15 10. Data management and analysis 17 11. Taking design seriously 17 © 2000 Statistical Services Centre, The University of Reading, UK 1. Introduction This guide describes the main concepts that are involved in designing an experiment. Its direct use is to assist scientists involved in the design of on-station field trials, but many of the concepts also apply to the design of other research exercises. These range from simulation exercises, and laboratory studies, to on-farm experiments, participatory studies and surveys. What characterises the design of on-station experiments is that the researcher has control of the treatments to apply and the units (plots) to which they will be applied. The results therefore usually provide a detailed knowledge of the effects of the treat- ments within the experiment. The major limitation is that they provide this information within the artificial "environment" of small plots in a research station. A laboratory study should achieve more precision, but in a yet more artificial environment. Even more precise is a simulation model, partly because it is then very cheap to collect data. -

The Theory of the Design of Experiments
The Theory of the Design of Experiments D.R. COX Honorary Fellow Nuffield College Oxford, UK AND N. REID Professor of Statistics University of Toronto, Canada CHAPMAN & HALL/CRC Boca Raton London New York Washington, D.C. C195X/disclaimer Page 1 Friday, April 28, 2000 10:59 AM Library of Congress Cataloging-in-Publication Data Cox, D. R. (David Roxbee) The theory of the design of experiments / D. R. Cox, N. Reid. p. cm. — (Monographs on statistics and applied probability ; 86) Includes bibliographical references and index. ISBN 1-58488-195-X (alk. paper) 1. Experimental design. I. Reid, N. II.Title. III. Series. QA279 .C73 2000 001.4 '34 —dc21 00-029529 CIP This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the author and the publisher cannot assume responsibility for the validity of all materials or for the consequences of their use. Neither this book nor any part may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage or retrieval system, without prior permission in writing from the publisher. The consent of CRC Press LLC does not extend to copying for general distribution, for promotion, for creating new works, or for resale. Specific permission must be obtained in writing from CRC Press LLC for such copying. Direct all inquiries to CRC Press LLC, 2000 N.W. -

Statistical Theory
Statistical Theory Prof. Gesine Reinert November 23, 2009 Aim: To review and extend the main ideas in Statistical Inference, both from a frequentist viewpoint and from a Bayesian viewpoint. This course serves not only as background to other courses, but also it will provide a basis for developing novel inference methods when faced with a new situation which includes uncertainty. Inference here includes estimating parameters and testing hypotheses. Overview • Part 1: Frequentist Statistics { Chapter 1: Likelihood, sufficiency and ancillarity. The Factoriza- tion Theorem. Exponential family models. { Chapter 2: Point estimation. When is an estimator a good estima- tor? Covering bias and variance, information, efficiency. Methods of estimation: Maximum likelihood estimation, nuisance parame- ters and profile likelihood; method of moments estimation. Bias and variance approximations via the delta method. { Chapter 3: Hypothesis testing. Pure significance tests, signifi- cance level. Simple hypotheses, Neyman-Pearson Lemma. Tests for composite hypotheses. Sample size calculation. Uniformly most powerful tests, Wald tests, score tests, generalised likelihood ratio tests. Multiple tests, combining independent tests. { Chapter 4: Interval estimation. Confidence sets and their con- nection with hypothesis tests. Approximate confidence intervals. Prediction sets. { Chapter 5: Asymptotic theory. Consistency. Asymptotic nor- mality of maximum likelihood estimates, score tests. Chi-square approximation for generalised likelihood ratio tests. Likelihood confidence regions. Pseudo-likelihood tests. • Part 2: Bayesian Statistics { Chapter 6: Background. Interpretations of probability; the Bayesian paradigm: prior distribution, posterior distribution, predictive distribution, credible intervals. Nuisance parameters are easy. 1 { Chapter 7: Bayesian models. Sufficiency, exchangeability. De Finetti's Theorem and its intepretation in Bayesian statistics. { Chapter 8: Prior distributions. Conjugate priors. -

Argus: Interactive a Priori Power Analysis
© 2020 IEEE. This is the author’s version of the article that has been published in IEEE Transactions on Visualization and Computer Graphics. The final version of this record is available at: xx.xxxx/TVCG.201x.xxxxxxx/ Argus: Interactive a priori Power Analysis Xiaoyi Wang, Alexander Eiselmayer, Wendy E. Mackay, Kasper Hornbæk, Chat Wacharamanotham 50 Reading Time (Minutes) Replications: 3 Power history Power of the hypothesis Screen – Paper 1.0 40 1.0 Replications: 2 0.8 0.8 30 Pairwise differences 0.6 0.6 20 0.4 0.4 10 0 1 2 3 4 5 6 One_Column Two_Column One_Column Two_Column 0.2 Minutes with 95% confidence interval 0.2 Screen Paper Fatigue Effect Minutes 0.0 0.0 0 2 3 4 6 8 10 12 14 8 12 16 20 24 28 32 36 40 44 48 Fig. 1: Argus interface: (A) Expected-averages view helps users estimate the means of the dependent variables through interactive chart. (B) Confound sliders incorporate potential confounds, e.g., fatigue or practice effects. (C) Power trade-off view simulates data to calculate statistical power; and (D) Pairwise-difference view displays confidence intervals for mean differences, animated as a dance of intervals. (E) History view displays an interactive power history tree so users can quickly compare statistical power with previously explored configurations. Abstract— A key challenge HCI researchers face when designing a controlled experiment is choosing the appropriate number of participants, or sample size. A priori power analysis examines the relationships among multiple parameters, including the complexity associated with human participants, e.g., order and fatigue effects, to calculate the statistical power of a given experiment design. -

Chapter 5 Statistical Inference
Chapter 5 Statistical Inference CHAPTER OUTLINE Section 1 Why Do We Need Statistics? Section 2 Inference Using a Probability Model Section 3 Statistical Models Section 4 Data Collection Section 5 Some Basic Inferences In this chapter, we begin our discussion of statistical inference. Probability theory is primarily concerned with calculating various quantities associated with a probability model. This requires that we know what the correct probability model is. In applica- tions, this is often not the case, and the best we can say is that the correct probability measure to use is in a set of possible probability measures. We refer to this collection as the statistical model. So, in a sense, our uncertainty has increased; not only do we have the uncertainty associated with an outcome or response as described by a probability measure, but now we are also uncertain about what the probability measure is. Statistical inference is concerned with making statements or inferences about char- acteristics of the true underlying probability measure. Of course, these inferences must be based on some kind of information; the statistical model makes up part of it. Another important part of the information will be given by an observed outcome or response, which we refer to as the data. Inferences then take the form of various statements about the true underlying probability measure from which the data were obtained. These take a variety of forms, which we refer to as types of inferences. The role of this chapter is to introduce the basic concepts and ideas of statistical inference. The most prominent approaches to inference are discussed in Chapters 6, 7, and 8. -

Design of Experiments and Data Analysis,” 2012 Reliability and Maintainability Symposium, January, 2012
Copyright © 2012 IEEE. Reprinted, with permission, from Huairui Guo and Adamantios Mettas, “Design of Experiments and Data Analysis,” 2012 Reliability and Maintainability Symposium, January, 2012. This material is posted here with permission of the IEEE. Such permission of the IEEE does not in any way imply IEEE endorsement of any of ReliaSoft Corporation's products or services. Internal or personal use of this material is permitted. However, permission to reprint/republish this material for advertising or promotional purposes or for creating new collective works for resale or redistribution must be obtained from the IEEE by writing to [email protected]. By choosing to view this document, you agree to all provisions of the copyright laws protecting it. 2012 Annual RELIABILITY and MAINTAINABILITY Symposium Design of Experiments and Data Analysis Huairui Guo, Ph. D. & Adamantios Mettas Huairui Guo, Ph.D., CPR. Adamantios Mettas, CPR ReliaSoft Corporation ReliaSoft Corporation 1450 S. Eastside Loop 1450 S. Eastside Loop Tucson, AZ 85710 USA Tucson, AZ 85710 USA e-mail: [email protected] e-mail: [email protected] Tutorial Notes © 2012 AR&MS SUMMARY & PURPOSE Design of Experiments (DOE) is one of the most useful statistical tools in product design and testing. While many organizations benefit from designed experiments, others are getting data with little useful information and wasting resources because of experiments that have not been carefully designed. Design of Experiments can be applied in many areas including but not limited to: design comparisons, variable identification, design optimization, process control and product performance prediction. Different design types in DOE have been developed for different purposes.