Extrasynaptic GABA-A Receptors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
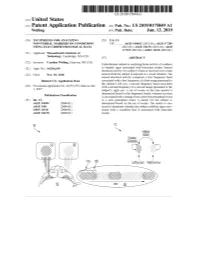
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

GABAA Transmission Is a Critical Step in the Process of Triggering Homeostatic Increases in Quantal Amplitude
GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude Jennifer C. Wilhelm and Peter Wenner* Department of Physiology, Emory University School of Medicine, Atlanta, GA 30322 Communicated by Lynn T. Landmesser, Case Western Reserve University, Cleveland, OH, June 23, 2008 (received for review March 1, 2008) When activity levels are altered over days, a network of cells is models suggests that reducing network activity produces a corre- capable of recognizing this perturbation and triggering several dis- sponding reduction in cellular spiking activity, thereby reducing tinct compensatory changes that should help to recover and maintain intracellular calcium levels in a postsynaptic cell. In this model, the original activity levels homeostatically. One feature commonly which we will refer to as the cell activity model, the postsynaptic cell observed after activity blockade has been a compensatory increase in senses changes in intracellular calcium levels as a measure of altered excitatory quantal amplitude. The sensing machinery that detects activity and triggers compensatory changes in mPSC amplitude. altered activity levels is a central focus of the field currently, but thus However, the studies that have inspired this model not only block far it has been elusive. The vast majority of studies that reduce spiking activity, they also block or reduce neurotransmitter binding network activity also reduce neurotransmission. We address the to its receptor and any associated downstream signaling cascades. possibility that reduced neurotransmission can trigger increases in Thus, it remains possible that neurotransmission is a critical step in quantal amplitude. In this work, we blocked glutamatergic or GABAA the sensing process that triggers changes in quantal amplitude. -

Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects
Neuropsychopharmacology (2005) 30, 833–841 & 2005 Nature Publishing Group All rights reserved 0893-133X/05 $30.00 www.neuropsychopharmacology.org Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects 1 2 ,3 1 Stefan Mathias , Josef Zihl , Axel Steiger* and Marike Lancel 1Section of Sleep Pharmacology, Max-Planck-Institute of Psychiatry, Munich, Germany; 2Section of Neuropsychology, Max-Planck-Institute of Psychiatry, Munich, Germany; 3Department of Psychiatry, Max-Planck-Institute of Psychiatry, Munich, Germany Aging is associated with dramatic reductions in sleep continuity and sleep intensity. Since gaboxadol, a selective GABAA receptor agonist, has been demonstrated to improve sleep consolidation and promote deep sleep, it may be an effective hypnotic, particularly for elderly patients with insomnia. In the present study, we investigated the effects of subchronic gaboxadol administration on nocturnal sleep and its residual effects during the next days in elderly subjects. This was a randomized, double-blind, placebo-controlled, balanced crossover study in 10 healthy elderly subjects without sleep complaints. The subjects were administered either placebo or 15 mg gaboxadol hydrochloride at bedtime on three consecutive nights. Sleep was recorded during each night from 2300 to 0700 h and tests assessing attention (target detection, stroop test) and memory function (visual form recognition, immediate word recall, digit span) were applied at 0900, 1400, and 1700 h during the following days. Compared with placebo, gaboxadol significantly shortened subjective sleep onset latency and increased self-rated sleep intensity and quality. Polysomnographic recordings showed that it significantly decreased the number of awakenings, the amount of intermittent wakefulness, and stage 1, and increased slow wave sleep and stage 2. -

Neurotransmitters-Drugs Andbrain Function.Pdf
Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Neurotransmitters, Drugs and Brain Function Edited by R. A. Webster Department of Pharmacology, University College London, UK JOHN WILEY & SONS, LTD Chichester Á New York Á Weinheim Á Brisbane Á Singapore Á Toronto Neurotransmitters, Drugs and Brain Function. Edited by Roy Webster Copyright & 2001 John Wiley & Sons Ltd ISBN: Hardback 0-471-97819-1 Paperback 0-471-98586-4 Electronic 0-470-84657-7 Copyright # 2001 by John Wiley & Sons Ltd. Bans Lane, Chichester, West Sussex PO19 1UD, UK National 01243 779777 International ++44) 1243 779777 e-mail +for orders and customer service enquiries): [email protected] Visit our Home Page on: http://www.wiley.co.uk or http://www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1P0LP,UK, without the permission in writing of the publisher. Other Wiley Editorial Oces John Wiley & Sons, Inc., 605 Third Avenue, New York, NY 10158-0012, USA WILEY-VCH Verlag GmbH, Pappelallee 3, D-69469 Weinheim, Germany John Wiley & Sons Australia, Ltd. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Gaboxadol Normalizes Behavioral Abnormalities in a Mouse Model of Fragile X Syndrome
ORIGINAL RESEARCH published: 25 June 2019 doi: 10.3389/fnbeh.2019.00141 Gaboxadol Normalizes Behavioral Abnormalities in a Mouse Model of Fragile X Syndrome Patricia Cogram 1,2,3,4, Robert M. J. Deacon 1,2,3,4, Jennifer L. Warner-Schmidt 5, Melanie J. von Schimmelmann 6, Brett S. Abrahams 6,7 and Matthew J. During 6,8* 1FRAXA-DVI, FRAXA Research Foundation, Boston, MA, United States, 2Centre for Systems Biotechnology, Biomedicine Division, Fraunhofer-Gesellschaft, Santiago, Chile, 3GEN.DDI Limited, London, United Kingdom, 4Institute of Ecology and Biodiversity (IEB), University of Chile, Santiago, Chile, 5NeuroJenic Consulting, LLC, Garden City, NY, United States, 6Ovid Therapeutics, New York, NY, United States, 7Department of Genetics and Neuroscience, Albert Einstein College of Medicine, Bronx, NY, United States, 8Department of Neurological Surgery and Molecular Virology, Immunology and Medical Genetics, Ohio State University College of Medicine, Columbus, OH, United States Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and autism. FXS is also accompanied by attention problems, hyperactivity, anxiety, aggression, poor sleep, repetitive behaviors, and self-injury. Recent work supports the role of g-aminobutyric-acid (GABA), the primary inhibitory neurotransmitter in the brain, in mediating symptoms of FXS. Deficits in GABA machinery have been observed in a mouse model of FXS, including a loss of tonic inhibition in the amygdala, which is Edited by: Martine Ammassari-Teule, mediated by extrasynaptic GABAA receptors. Humans with FXS also show reduced Italian National Research Council GABAA receptor availability. Here, we sought to evaluate the potential of gaboxadol (CNR), Italy (also called OV101 and THIP), a selective and potent agonist for delta-subunit-containing Reviewed by: extrasynaptic GABA receptors (dSEGA), as a therapeutic agent for FXS by assessing Giulia Poggi, A University of Zurich, Switzerland its ability to normalize aberrant behaviors in a relatively uncharacterized mouse model Valerie J. -

Ligand-Gated Ion Channels' British Journal of Pharmacology, Vol
Edinburgh Research Explorer The Concise Guide to PHARMACOLOGY 2015/16 Citation for published version: Alexander, SP, Peters, JA, Kelly, E, Marrion, N, Benson, HE, Faccenda, E, Pawson, AJ, Sharman, JL, Southan, C, Davies, JA & CGTP Collaborators 2015, 'The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels' British Journal of Pharmacology, vol. 172, no. 24, pp. 5870-5903. DOI: 10.1111/bph.13350 Digital Object Identifier (DOI): 10.1111/bph.13350 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 05. Apr. 2019 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels. British Journal of Pharmacology (2015) 172, 5870–5903 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Ligand-gated ion channels Stephen PH Alexander1, -

Cell Surface Mobility of GABAB Receptors Saad Bin
Cell surface mobility of GABAB receptors Saad Bin Hannan September 2011 A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy of the University College London Department of Neuroscience, Physiology, and Pharmacology University College London Gower Street London WC1E 6BT UK Declaration ii ‘I, Saad Hannan confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis.' ____________________ Saad Hannan September 2011 To Ammu, Abbu, Polu Abstract ivi Abstract Type-B γ-aminobutyric acid receptors (GABABRs) are important for mediating slow inhibition in the central nervous system and the kinetics of their internalisation and lateral mobility will be a major determinant of their signalling efficacy. Functional GABABRs require R1 and R2 subunit co-assembly, but how heterodimerisation affects the trafficking kinetics of GABABRs is unknown. Here, an α- bungarotoxin binding site (BBS) was inserted into the N-terminus of R2 to monitor receptor mobility in live cells. GABABRs are internalised via clathrin- and dynamin- dependent pathways and recruited to endosomes. By mutating the BBS, a new technique was developed to differentially track R1a and R2 simultaneously, revealing the subunits internalise as heteromers and that R2 dominantly-affects constitutive internalisation of GABABRs. Notably, the internalisation profile of R1aR2 heteromers, but not R1a homomers devoid of their ER retention motif (R1ASA), is similar to R2 homomers in heterologous systems. The internalisation of R1aASA was slowed to that of R2 by mutating a di-leucine motif in the R1 C-terminus, indicating a new role for heterodimerisation, whereby R2 subunits slow the internalization of surface GABABRs. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Isoguvacine Hydrochloride | Medchemexpress
Inhibitors Product Data Sheet Isoguvacine hydrochloride • Agonists Cat. No.: HY-100810 CAS No.: 68547-97-7 Molecular Formula: C₆H₁₀ClNO₂ • Molecular Weight: 163.6 Screening Libraries Target: GABA Receptor Pathway: Membrane Transporter/Ion Channel; Neuronal Signaling Storage: Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro H2O : ≥ 100 mg/mL (611.25 mM) DMSO : 25 mg/mL (152.81 mM; Need ultrasonic) * "≥" means soluble, but saturation unknown. Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 6.1125 mL 30.5623 mL 61.1247 mL Stock Solutions 5 mM 1.2225 mL 6.1125 mL 12.2249 mL 10 mM 0.6112 mL 3.0562 mL 6.1125 mL Please refer to the solubility information to select the appropriate solvent. In Vivo 1. Add each solvent one by one: 10% DMSO >> 40% PEG300 >> 5% Tween-80 >> 45% saline Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution 2. Add each solvent one by one: 10% DMSO >> 90% (20% SBE-β-CD in saline) Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution 3. Add each solvent one by one: 10% DMSO >> 90% corn oil Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution BIOLOGICAL ACTIVITY Description Isoguvacine hydrochloride is a GABA receptor agonist. IC₅₀ & Target GABA[1] In Vitro Isoguvacine binds to a mouse forebrain synaptic membrane preparation. The specific binding is displaceable by GABA, Page 1 of 2 www.MedChemExpress.com muscimol and bicuculline but not by picrotoxin or diaminobutyric acid. -

Gabaergic Signaling Linked to Autophagy Enhances Host Protection Against Intracellular Bacterial Infections
ARTICLE DOI: 10.1038/s41467-018-06487-5 OPEN GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections Jin Kyung Kim1,2,3, Yi Sak Kim1,2,3, Hye-Mi Lee1,3, Hyo Sun Jin4, Chiranjivi Neupane 2,5, Sup Kim1,2,3, Sang-Hee Lee6, Jung-Joon Min7, Miwa Sasai8, Jae-Ho Jeong 9,10, Seong-Kyu Choe11, Jin-Man Kim12, Masahiro Yamamoto8, Hyon E. Choy 9,10, Jin Bong Park 2,5 & Eun-Kyeong Jo1,2,3 1234567890():,; Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter in the brain; however, the roles of GABA in antimicrobial host defenses are largely unknown. Here we demonstrate that GABAergic activation enhances antimicrobial responses against intracel- lular bacterial infection. Intracellular bacterial infection decreases GABA levels in vitro in macrophages and in vivo in sera. Treatment of macrophages with GABA or GABAergic drugs promotes autophagy activation, enhances phagosomal maturation and antimicrobial responses against mycobacterial infection. In macrophages, the GABAergic defense is mediated via macrophage type A GABA receptor (GABAAR), intracellular calcium release, and the GABA type A receptor-associated protein-like 1 (GABARAPL1; an Atg8 homolog). Finally, GABAergic inhibition increases bacterial loads in mice and zebrafish in vivo, sug- gesting that the GABAergic defense plays an essential function in metazoan host defenses. Our study identified a previously unappreciated role for GABAergic signaling in linking antibacterial autophagy to enhance host innate defense against intracellular bacterial infection. 1 Department of Microbiology, Chungnam National University School of Medicine, Daejeon 35015, Korea. 2 Department of Medical Science, Chungnam National University School of Medicine, Daejeon 35015, Korea. -

Bicuculline and Gabazine Are Allosteric Inhibitors of Channel Opening of the GABAA Receptor
The Journal of Neuroscience, January 15, 1997, 17(2):625–634 Bicuculline and Gabazine Are Allosteric Inhibitors of Channel Opening of the GABAA Receptor Shinya Ueno,1 John Bracamontes,1 Chuck Zorumski,2 David S. Weiss,3 and Joe Henry Steinbach1 Departments of 1Anesthesiology and 2Psychiatry, Washington University School of Medicine, St. Louis, Missouri 63110, and 3University of Alabama at Birmingham, Neurobiology Research Center and Department of Physiology and Biophysics, Birmingham, Alabama 35294-0021 Anesthetic drugs are known to interact with GABAA receptors, bicuculline only partially blocked responses to pentobarbital. both to potentiate the effects of low concentrations of GABA and These observations indicate that the blockers do not compete to directly gate open the ion channel in the absence of GABA; with alphaxalone or pentobarbital for a single class of sites on the however, the site(s) involved in direct gating by these drugs is not GABAA receptor. Finally, at receptors containing a1b2(Y157S)g2L known. We have studied the ability of alphaxalone (an anesthetic subunits, both bicuculline and gabazine showed weak agonist steroid) and pentobarbital (an anesthetic barbiturate) to directly activity and actually potentiated responses to alphaxalone. These activate recombinant GABAA receptors containing the a1, b2, and observations indicate that the blocking drugs can produce allo- g2L subunits. Steroid gating was not affected when either of two steric changes in GABAA receptors, at least those containing this mutated b2 subunits [b2(Y157S) and b2(Y205S)] are incorporated mutated b2 subunit. We conclude that the sites for binding ste- into the receptors, although these subunits greatly reduce the roids and barbiturates do not overlap with the GABA-binding site.