Practical Aspects on How to Deracemize Atropizomers by Means of Crystallization Ryusei Oketani
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chiral Auxiliaries and Optical Resolving Agents
Chiral Auxiliaries and Optical Resolving Agents Most bioactive substances are optically active. For instance, if This brochure introduces a variety of chiral auxiliaries and a substance is synthesized as a racemic compound, its optical resolving agents. We hope that it will be useful for your enantiomer may show no activity or even undesired bioactivity. research of the synthesis of optically active compounds. Thus, methods to gain enantiopure compounds have been Additionally, TCI has some brochures introducing chiral developed. When synthesizing enantiopure compounds, the compounds for the chiral pool method in “Chiral Building Blocks”, methods are roughly divided into three methods. “Terpenes”, “Amino Acids” and other brochures. Sugar derivatives are also introduced in a catalog, “Reagents for Glyco Chemistry Chiral pool method: & Biology”, and category pages of sugar chains. Furthermore, The method using an easily available chiral compound as a TCI has many kinds of catalysts for asymmetric synthesis and starting material like an amino acid or sugar. introduce them in brochures such as “Asymmetric Synthesis” and Asymmetric synthesis: “Asymmetric Organocatalysts”, and other contents. The method to introduce an asymmetric point to compounds You can search our information through “asymmetric synthesis” without an asymmetric point. Syntheses using achiral as a keyword. auxiliaries are included here. Optical resolution: The method to separate a racemic compound into two ● Reactions with Chiral Auxiliaries enantiomers. The direct method using a chiral column and One of the most famous named reactions using chiral auxiliaries1) the indirect method to separate two enantiomers using is the Evans aldol reaction.2) This reaction is quite useful because optical resolving agents to convert into diastereomers are this reaction can efficiently introduce two asymmetric carbons into examples. -

Lectures 2014
Selective Organic Synthesis KD 2390 (9 hp) Christina Moberg The advent of organic chemistry shaped the world • Biology is dependent on organic synthesis • Organic synthesis is the foundation for biotechnology, nanotechnology, pharmaceutical industry, and material industry About the course: • Disposition: lectures, exercises, lab, workshop • Course book: Clayden, Greeves and Warren Organic Chemistry, Oxford University Press, 2012 (ISBN 978-0-19-927029-3) [or Clayden, Greeves, Warren and Wothers: Organic Chemistry, Oxford University Press, 2001 (ISBN 0 19 850346 6)] and distributed material • Examination: oral exam, lab work (lab and workshop compulsory) After the course the student should be able to:" " • Describe basic stereochemical concepts • Describe principles for stereoselective synthesis, in particular for enantioselective synthesis • Explain the stereochemistry observed in chemical reactions • Suggest methods for stereoselective synthesis of simple organic compounds containing stereogenic elements • Identify suitable reagents for stereoselective transformations • Use retrosynthetic analysis for the construction of synthetic routes for simple organic compounds • Prepare organic compounds using advanced synthetic methodology Contents • Fundamental stereochemical concepts • Synthetic strategy and principles for stereoselective, in particular enantioselective, chemical transformations • Transition metal catalysis • Applications of frontier orbital theory • Retrosynthetic analysis • Advanced organic synthesis Marcellin Pierre Eugène Berthelot (1827 - 1907) " "La Chimie crée ses objets" “This is an important point: neither biology nor chemistry would be served best by a development in which all organic chemists would simply become biological such that, as a consequence, research at the core of organic chemistry and, therefore, progress in understanding the reactivity of organic molecules, would dry out. Progress at its core in understanding and reasoning in not only essential for organic chemistry itself, but for life sciences as a whole. -

Enzyme Supported Crystallization of Chiral Amino Acids
ISBN 978-3-89336-715-3 40 Band /Volume Gesundheit /Health 40 Gesundheit Enzyme supported crystallization Health Kerstin Würges of chiral amino acids Mitglied der Helmholtz-Gemeinschaft Kerstin Würges Kerstin aminoacids Enzyme supported ofchiral crystallization Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 Forschungszentrum Jülich GmbH Institute of Bio- and Geosciences (IBG) Biotechnology (IBG-1) Enzyme supported crystallization of chiral amino acids Kerstin Würges Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 ISSN 1866-1785 ISBN 978-3-89336-715-3 Bibliographic information published by the Deutsche Nationalbibliothek. The Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografie; detailed bibliographic data are available in the Internet at http://dnb.d-nb.de. Publisher and Forschungszentrum Jülich GmbH Distributor: Zentralbibliothek 52425 Jülich Phone +49 (0) 24 61 61-53 68 · Fax +49 (0) 24 61 61-61 03 e-mail: [email protected] Internet: http://www.fz-juelich.de/zb Cover Design: Grafische Medien, Forschungszentrum Jülich GmbH Printer: Grafische Medien, Forschungszentrum Jülich GmbH Copyright: Forschungszentrum Jülich 2011 Schriften des Forschungszentrums Jülich Reihe Gesundheit / Health Band / Volume 40 D 61 (Diss. Düsseldorf, Univ., 2011) ISSN 1866-1785 ISBN 978-3-89336-715-3 The complete volume ist freely available on the Internet on the Jülicher Open Access Server (JUWEL) at http://www.fz-juelich.de/zb/juwel Neither this book nor any part of it may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage and retrieval system, without permission in writing from the publisher. -

Characterization of Stereoselective Metabolism, Inhibitory Effect
DMD Fast Forward. Published on November 15, 2018 as DOI: 10.1124/dmd.118.080549 This article has not been copyedited and formatted. The final version may differ from this version. Journal Drug Metabolism and Disposition CHARACTERIZATION OF STEREOSELECTIVE METABOLISM, INHIBITORY EFFECT ON URIC ACID UPTAKE TRANSPORTERS, AND PHARMACOKINETICS OF LESINURAD ATROPISOMERS Downloaded from Chun Yang, Dongmei Zhou, Zancong Shen, David M. Wilson, Matthew Renner, Jeffrey N. Miner, Jean-Luc Girardet and Caroline A. Lee dmd.aspetjournals.org Preclinical & Clinical DMPK (CY, ZS, CAL), Bioanalytical (DZ, DMW), Biology (JM) and Chemistry (J-LG, MR) Ardea Biosciences, Inc., San Diego, CA at ASPET Journals on October 1, 2021 1 DMD Fast Forward. Published on November 15, 2018 as DOI: 10.1124/dmd.118.080549 This article has not been copyedited and formatted. The final version may differ from this version. Running Title: Lesinurad Atropisomer Characterization Corresponding Author: Caroline A. Lee, PhD Downloaded from Translational Sciences, Ardea Biosciences 9390 Towne Centre Drive dmd.aspetjournals.org San Diego, CA 92121 Email: [email protected] at ASPET Journals on October 1, 2021 Tel: 858-449-1797 Manuscript Format Abstract: 223 words Introduction: 599 words Discussion: 1408 words Number of Text Pages: 29 2 DMD Fast Forward. Published on November 15, 2018 as DOI: 10.1124/dmd.118.080549 This article has not been copyedited and formatted. The final version may differ from this version. References: 21 Number of Tables: 2 Number of Figures: 8 Supplemental Table: 0 Supplemental Figures: 0 Downloaded from dmd.aspetjournals.org at ASPET Journals on October 1, 2021 3 DMD Fast Forward. -

Catalytic Asymmetric Synthesis
Catalytic Asymmetric Synthesis Dr Alan Armstrong Rm 313 (RCS1), ext. 45876; [email protected] 7 lectures Recommended texts: “Catalytic Asymmetric Synthesis, 2nd edition”, ed. I Ojima, Wiley-VCH, 2000 (or 1st ed., 1993, which contains most of the lecture material) “Asymmetric Synthesis”, G Procter, Oxford, 1996 Aims of course: To give students an understanding of the basic principles of asymmetric catalysis, and to demonstrate these in the context of state-of-the-art catalytic asymmetric processes for C-C bond formation and redox processes. Course objectives: At the end of this course you should be able to: • Recognise the types of functional groups which can be prepared by catalytic asymmetric methods discussed in the course; • Use this knowledge in planning the synthesis of enantiomerically enriched compounds from given prochiral starting materials; • Outline the scope and limitations of any methods you propose, with respect to parameters such as turnover, substrate and functional group tolerance, availability of catalysts and/or ligands etc Course content (by lecture, approximately): 1. Introduction and general principles of asymmetric catalysis. Oxidation of functionalised olefins: asymmetric epoxidation of allylic alcohols and enones 2. Oxidation of unfunctionalised olefins: epoxidation, dihydroxylation and aminohydroxylation. 3. Reduction of olefins: asymmetric hydrogenation. 4. Reduction of ketones and imines 5. Asymmetric C-C bond formation: nucleophilic attack on carbonyls, imines and enones 6. Asymmetric transition metal catalysis in C-C bond forming reactions (cross-couplings, Heck reactions, allylic substitution, carbenoid reactions) 7. Chiral Lewis acid catalysis (aldol reactions, cycloadditions, Mannich reactions, allylations) 1 Catalytic Asymmetric Synthesis - Lecture 1 Background and general principles • Why asymmetric synthesis? The need to prepare pharmaceuticals and other fine chemicals as single enantiomers drives the field of asymmetric synthesis. -

Stereochemistry of Bruice's Organic Chemistry Before Embarking on Comprehending This Summary
Enantioselective synthesis It helps if you review the concepts in Chapter 5: Stereochemistry of Bruice's Organic Chemistry before embarking on comprehending this summary Most of the molecules on earth are chiral, such as amino acids and carbohydrates. Enantioselective synthesis is an important means by which enantiopure chiral molecules may be obtained for study and medicine. The study of enantioselective synthesis has evolved from issues of diastereoselectivity, through auxiliary-based methods for the synthesis of enantiomerically pure compounds (diastereoselectivity followed by separation and auxiliary cleavage), to asymmetric catalysis. In the latter technology, enantiomers are the products, and highly selective reactions allow preparation - in a single step - of chiral substances in high enantiomeric excess (ee) for many reaction types. Enantioselective synthesis, also known as asymmetric synthesis, is organic synthesis that introduces one or more new and desired elements of chirality. This methodology is important in the field of pharmaceuticals because the different enantiomers or diastereomers of a molecule often have different biological activity. See examples from Chapter 5 of Bruice's Organic Chemistry. Methodologies of Asymmetric Synthesis Chiral pool synthesis Chiral pool synthesis is the easiest approach: A chiral starting material is manipulated through successive reactions using achiral reagents that retain its chirality to obtain the desired target molecule. This is especially attractive for target molecules having the similar chirality to a relatively inexpensive naturally occurring building-block such as a sugar or amino acid. However, the number of possible reactions the molecule can undergo is restricted, and lengthy synthetic routes may be required. Also, this approach requires a stoichiometric amount of the enantiopure starting material, which may be rather expensive if not occurring in nature, whereas chiral catalysis requires only a catalytic amount of chiral material. -

Atropisomerism
Atropisomerism Metal-mediated transformations to set biaryl chirality Nathaniel Kadunce Literature Meeting 12 December 2014 Atropisomerism • Atropos- the “inflexible” or “without turn” • Arise from hindered rotation about a single bond allowing for isolation of separate conformers HO2C NO2 HO2C NO2 HO2C NO2 HO2C NO2 Ra Sa 6,6'-dinitro-2,2'-diphenic acid, the first experimentally described atropisomeric compound Journal of the Chemical Society, Transactions 121: 614 First Evidence • Kaufler Hypothesis: cis and trans isomers explain op9cal proper9es • Supported by a series of misassignments in derivave studies • “It has been suggested by Carothers and by Mascarrelli that an objec9on to the Kaufler formula which has never been emphasized is the necessity of bending a bond to an angle of 90°. With a Kekule nucleus such a formula cannot be constructed, and each 9me that chemists adobt assump9ons that cannot be reconciled with the Kekule nucleus they encounter difficul9es.” –Adams, 1933 Rogers, A. and Yuan. H. C. Chem. Rev. 1933, 12, 261. First Evidence • Kaufler Hypothesis: cis and trans isomers explain op9cal proper9es • Supported by a series of misassignments in derivave studies • “It has been suggested by Carothers and by Mascarrelli that an objec9on to the Kaufler formula which has never been emphasized is the necessity of bending a bond to an angle of 90°. With a Kekule nucleus such a formula cannot be constructed, and each 9me that chemists adobt assump9ons that cannot be reconciled with the Kekule nucleus they encounter difficul9es.” –Adams, 1933 -

Asymmetric Synthesis
Chapter 45 — Asymmetric synthesis - Pure enantiomers from Nature: the chiral pool and chiral induction - Asymmetric synthesis: chiral auxiliaries Enolate alkylation Aldol reaction - Enantiomeric excess (ee) - Asymmetric synthesis: chiral reagents and catalysts CBS reagent for chiral reductions Sharpless asymmetric epoxidation Synthesizing pure enantiomers starting from Nature’s chiral pool AcO OH HO B O O HO – O O HO + O N OH H3N OH HO OH O H … AcO HO OH O OH Sulcatol - insect pheremone HO Synthesis from the chiral pool — chiral induction turns one stereocenter into many 40 steps Me HO H Me H O O OH O O OBn O * * Only one H O HO chiral reagent H H Me OBn Me Deoxyribose Fragment of Brevetoxin B Asymmetric synthesis 1. Producing a new stereogenic centre on an achiral molecule makes two enantiomeric transition states of equal energy… and therefore two enantiomeric products in equal amounts. O O 1 1 Nu R R2 R R2 Nu E OH OH 1 O 1 Nu R R Nu R2 R2 1 R R2 O Ph OH HO Ph PhLi + N N N Asymmetric synthesis 2. O 1 R R* Nu When there is an existing O chiral centre, the two possible TS’s are diastereomeric and E 1 Nu R R* can be of different energy. Thus one isomer of the new stereogenic centre can be OH O OH produced in a larger amount. 1 1 R Nu Nu R *R 1 R* R R* HO Ph O O O Ph OH PhLi PhLi Ph Ph N N N N N Ph N N N N N OH Ph O O O Ph OH Ph A removable chiral centre… synthesis with chiral auxiliaries O ??? O R R 1) Add a chiral 3) Remove the auxiliary chiral auxiliary O O R* R* 2) Add the new stereocentre via chiral induction Enantiopure oxazolidinones as chiral auxiliaries 1. -

Enantiomeric Chromatographic Separations and Ionic Liquids Jie Ding Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2005 Enantiomeric chromatographic separations and ionic liquids Jie Ding Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons, Medicinal and Pharmaceutical Chemistry Commons, Medicinal Chemistry and Pharmaceutics Commons, Medicinal-Pharmaceutical Chemistry Commons, and the Organic Chemistry Commons Recommended Citation Ding, Jie, "Enantiomeric chromatographic separations and ionic liquids " (2005). Retrospective Theses and Dissertations. 1725. https://lib.dr.iastate.edu/rtd/1725 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Enantiomeric chromatographic separations and ionic liquids by Jie Ding A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Analytical Chemistry Program of Study Committee: Daniel W. Armstrong, Major Professor Lee Anne Willson Robert S. Houk Jacob W. Petrich Richard C. Larock Iowa State University Ames, Iowa 2005 Copyright © Jie Ding, 2005. All rights reserved. UMI Number: 3200412 Copyright 2005 by Ding, Jie All rights reserved. INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Atropisomerism: Axial Chirality in Nature and Synthesis
R R * R MLn Me R OH O OH N OMe * Me Me Atropisomerism O * MeO OH N Axial Chirality in Nature and Synthesis H OMe Me MeO O X Y Y X OH O Y X X Y OMe MeO OH Kevin Allan O H MeO * OH Stoltz Group Literature Meeting Al Li Monday 11/14/2005 * O OEt 8 pm OMe 147 Noyes OH O O O O Nu O Nu Outline I. Introduction a. History and Background b. Conditions for Axial Chirality II. Direct Atroposelective Aryl-Aryl Coupling a. Diastereoselective Coupling b. Enantioselective Coupling III. Resolution/Desymmetrization of Prostereogenic Biaryls a. Configurationally Stable, Axially Achiral b. Configurationally Unstable, Axially Chiral Motokazu Uemura (right) and i. Atropodiastereoselective Bridge Formation Ken Kamikawa (left) ii. Atroposelective Bridge Cleavage / The "Lactone Method" Osaka Prefecture University IV. Atroposelective Construction of an Aromatic Ring Albert I. Meyers Colorado State University For a general review of biaryls in synthesis: Bringmann, G.; Mortimer, A. J. P.; Keller, P. A.; Gresser, M. J.; Garner, J.; Breuning, M. Angew. Chem. I. E. 2005, 44, 5384-5427. Biaryl cross-coupling: Stanforth, S. P. Tetrahedron. 1998, 54, 263-303. Lloyd-Williams, P.; Giralt, E. Chem. Soc. Rev. 2001, 30, 145-157. Broutin, P.-E.; Colobert, F. Eur. J. Org. Chem. 2005, 1113-1128. Lactone Methodology: Gerhard Bringmann Matthias Breuning Bringmann, G.; Breuning, M.; Tasler, S. Synthesis, 1999, 4, 525-558. Universitat Wurzburg Universitat Wurzburg Bringmann, G.; Tasler, S.; Pfeifer, R.-M.; Breuning, M. J. Organomet. Chem. 2002, 661, 49-65. Atropisomerism is Everywhere -

Stereochemistry the Different Types of Isomers. Stereochemistry Focuses
Stereochemistry The different types of isomers. Stereochemistry focuses on stereoisomers Stereochemistry, a subdiscipline of chemistry, involves the study of the relative spatial arrangement of atoms that form the structure of molecules and their manipulation. An important branch of stereochemistry is the study of chiral molecules. Stereochemistry is also known as 3D chemistry because the prefix "stereo-" means "three- dimensionality". The study of stereochemistry focuses on stereoisomers and spans the entire spectrum of organic, inorganic, biological, physical and especially supramolecular chemistry. Stereochemistry includes methods for determining and describing these relationships; the effect on the physical or biological properties these relationships impart upon the molecules in question, and the manner in which these relationships influence the reactivity of the molecules in question (dynamic stereochemistry). History Louis Pasteur could rightly be described as the first stereochemist, having observed in 1849 that salts of tartaric acid collected from wine production vessels could rotate plane polarized light, but that salts from other sources did not. This property, the only physical property in which the two types of tartrate salts differed, is due to optical isomerism. In 1874, Jacobus Henricus van 't Hoff and Joseph Le Belexplained optical activity in terms of the tetrahedral arrangement of the atoms bound to carbon. Significance Cahn–Ingold–Prelog priority rules are part of a system for describing a molecule's stereochemistry. They rank the atoms around a stereocenter in a standard way, allowing the relative position of these atoms in the molecule to be described unambiguously. A Fischer projection is a simplified way to depict the stereochemistry around a stereocenter. -
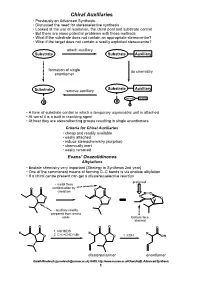
Chiral Auxiliaries • Previously on Advanced Synthesis
Chiral Auxiliaries • Previously on Advanced Synthesis..... • Discussed the need for stereoselective synthesis • Looked at the use of resolution, the chiral pool and substrate control • But there are some potential problems with those methods • What if the substrate does not contain an appropriate stereocentre? • What if the target does not contain a readily exploited stereocentre? attach auxiliary Substrate Substrate Auxiliary formation of single do chemistry enantiomer Substrate Auxiliary Substrate remove auxiliary Substrate Auxiliary R R R • A form of substrate control in which a temporary asymmetric unit is attached • At worst it is a built in resolving agent • At best they are stereodirecting groups resulting in single enantiomers Criteria for Chiral Auxiliaries • cheap and readily available • easily attached • induce stereochemistry (surprise) • chemically inert • easily removed Evans' Oxazolidinones Alkylations • Enolate chemistry very important (Strategy in Synthesis 2nd year) • One of the commonest means of forming C–C bonds is via enolate alkylation • If a chiral centre present can get a diastereoselective reaction preferred • metal fixes M O O conformation by O chelation O M O N O O O N O N H • auxiliary readily prepared from amino acids Bottom face blocked O O O O O 1. NaHMDS R R R N O 2. CH2=CHCH2Br N O 1. KOH OH diastereoisomer enantiomer Gareth Rowlands ([email protected]) Ar402, http://www.sussex.ac.uk/Users/kafj6, Advanced Synthesis 1 • The aldol reaction is an extremely valuable means of introducing stereochemistry