Data Mining the Genetics of Leukemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bacillus Anthracis' Lethal Toxin Induces Broad Transcriptional Responses In
Chauncey et al. BMC Immunology 2012, 13:33 http://www.biomedcentral.com/1471-2172/13/33 RESEARCH ARTICLE Open Access Bacillus anthracis’ lethal toxin induces broad transcriptional responses in human peripheral monocytes Kassidy M Chauncey1, M Cecilia Lopez2, Gurjit Sidhu1, Sarah E Szarowicz1, Henry V Baker2, Conrad Quinn3 and Frederick S Southwick1* Abstract Background: Anthrax lethal toxin (LT), produced by the Gram-positive bacterium Bacillus anthracis, is a highly effective zinc dependent metalloprotease that cleaves the N-terminus of mitogen-activated protein kinase kinases (MAPKK or MEKs) and is known to play a role in impairing the host immune system during an inhalation anthrax infection. Here, we present the transcriptional responses of LT treated human monocytes in order to further elucidate the mechanisms of LT inhibition on the host immune system. Results: Western Blot analysis demonstrated cleavage of endogenous MEK1 and MEK3 when human monocytes were treated with 500 ng/mL LT for four hours, proving their susceptibility to anthrax lethal toxin. Furthermore, staining with annexin V and propidium iodide revealed that LT treatment did not induce human peripheral monocyte apoptosis or necrosis. Using Affymetrix Human Genome U133 Plus 2.0 Arrays, we identified over 820 probe sets differentially regulated after LT treatment at the p <0.001 significance level, interrupting the normal transduction of over 60 known pathways. As expected, the MAPKK signaling pathway was most drastically affected by LT, but numerous genes outside the well-recognized pathways were also influenced by LT including the IL-18 signaling pathway, Toll-like receptor pathway and the IFN alpha signaling pathway. -

Unravelling the Cell Adhesion Defect in Meckel-Gruber Syndrome
Unravelling the Cell Adhesion Defect in Meckel-Gruber Syndrome Submitted by Benjamin Roland Alexander Meadows as a thesis for the degree of Doctor of Philosophy in Biological Sciences in September 2016 This thesis is available for Library use on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. I certify that all material in this thesis which is not my own work has been identified and that no material has previously been submitted and approved for the award of a degree by this or any other University. ……………………………………………………………… 1 2 Acknowledgements The first people who must be thanked are my fellow Dawe group members Kate McIntosh, Kat Curry, and half of Holly Hardy, as well as all past group members and Helen herself, who has always been a supportive and patient supervisor with a worryingly encyclopaedic knowledge of the human proteome. Some (in the end, distressingly small) parts of this project would not have been possible without my western blot consultancy team, including senior western blot consultant Joe Costello and junior consultants Afsoon Sadeghi-Azadi, Jack Chen, Luis Godinho, Tina Schrader, Stacey Scott, Lucy Green, and Lizzy Anderson. James Wakefield is thanked for improvising a protocol for actin co- sedimentation out of almost thin air. Most surprisingly, it worked. Special thanks are due to the many undergraduates who have contributed to this project, without whose hard work many an n would be low: Beth Hickton, Grace Howells, Annie Toynbee, Alex Oldfield, Leonie Hawksley, and Georgie McDonald. Peter Splatt and Christian Hacker are thanked for their help with electron microscopy. -
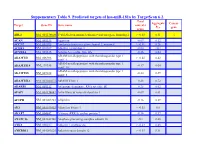
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Bacillus Anthracis' Lethal Toxin Induces Broad Transcriptional Responses In
Chauncey et al. BMC Immunology 2012, 13:33 http://www.biomedcentral.com/1471-2172/13/33 RESEARCH ARTICLE Open Access Bacillus anthracis’ lethal toxin induces broad transcriptional responses in human peripheral monocytes Kassidy M Chauncey1, M Cecilia Lopez2, Gurjit Sidhu1, Sarah E Szarowicz1, Henry V Baker2, Conrad Quinn3 and Frederick S Southwick1* Abstract Background: Anthrax lethal toxin (LT), produced by the Gram-positive bacterium Bacillus anthracis, is a highly effective zinc dependent metalloprotease that cleaves the N-terminus of mitogen-activated protein kinase kinases (MAPKK or MEKs) and is known to play a role in impairing the host immune system during an inhalation anthrax infection. Here, we present the transcriptional responses of LT treated human monocytes in order to further elucidate the mechanisms of LT inhibition on the host immune system. Results: Western Blot analysis demonstrated cleavage of endogenous MEK1 and MEK3 when human monocytes were treated with 500 ng/mL LT for four hours, proving their susceptibility to anthrax lethal toxin. Furthermore, staining with annexin V and propidium iodide revealed that LT treatment did not induce human peripheral monocyte apoptosis or necrosis. Using Affymetrix Human Genome U133 Plus 2.0 Arrays, we identified over 820 probe sets differentially regulated after LT treatment at the p <0.001 significance level, interrupting the normal transduction of over 60 known pathways. As expected, the MAPKK signaling pathway was most drastically affected by LT, but numerous genes outside the well-recognized pathways were also influenced by LT including the IL-18 signaling pathway, Toll-like receptor pathway and the IFN alpha signaling pathway. -

Biomarker Discovery for Asthma Phenotyping: from Gene Expression to the Clinic
UvA-DARE (Digital Academic Repository) Biomarker discovery for asthma phenotyping: From gene expression to the clinic Wagener, A.H. Publication date 2016 Document Version Final published version Link to publication Citation for published version (APA): Wagener, A. H. (2016). Biomarker discovery for asthma phenotyping: From gene expression to the clinic. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:25 Sep 2021 CHAPTER 3 Supporting Information File The impact of allergic rhinitis and asthma on human nasal and bronchial epithelial gene expression methods Primary epithelial cell culture Primary cells were obtained by first digesting the biopsies and brushings with collage- nase 4 (Worthington Biochemical Corp., Lakewood, NJ, USA) for 1 hour in Hanks’ bal- anced salt solution (Sigma-Aldrich, Zwijndrecht, The Netherlands). -

Identification of Conserved Genes Triggering Puberty in European Sea Bass Males ( Dicentrarchus Labrax ) by Microarray Expression Profiling
Retinoic acid signaling pathway: gene regulation during the onset of puberty in the European sea bass Ruta de señalización del ácido retinoico: regulación génica durante el inicio de la pubertad en la lubina Europea Paula Javiera Medina Henríquez Thesis presented to obtain the Ph.D. degree at the Polytechnic University of Catalonia (UPC) Ph. D. Program in Marine Sciences 2019 Thesis supervisor: Dra. Mercedes Blázquez Peinado (ICM-CSIC) Barcelona, 2019 Esta Tesis ha sido realizada en el Departamento de Recursos Marinos Renovables del Instituto de Ciencias del Mar de Barcelona (ICM-CSIC). Parte de la experimentación también se ha llevado a cabo en el Instituto de Acuicultura Torre de la sal (IATS-CSIC), a través de una estrecha colaboración con el grupo de Fisiología de la Reproducción de Peces. La financiación económica para la e xperimentación se ha recibido a través de los siguientes Proyectos de Investigación: Mejora de la producción en acuicultura mediante el uso de herramientas biotecnológicas CSD2007-00002 (AQUAGENOMICS). Bases moleculares, celulares y endocrinas de la pubertad y del desarrollo gonadal en lubina ( Dicentrarchus labrax ). Desarrollo de tecnologías ambientales, hormonales, y de manipulación genética para su control PROMETEO/2010/003 (REPROBASS). Regulación Hormonal y desarrollo del eje hipotalámico-hipofisario-gonadal durante la diferenciación sexual y la pubertad en peces teleósteos PIE 200930I037(TERMOBASS). Regulación endocrina y paracrina de la diferenciación sexual y el desarrollo gonadal en la lubina AGL2011-28890 (REPROSEX). Acción complementaria al proyecto: regulación endocrina y paracrina de la diferenciación sexual y desarrollo gonadal en la lubina (REPROSEX). Avances en el control de la pubertad y ciclo reproductor de la lubina y su aplicación tecnológica a otras especies de peces PROMETEO II/2014/051 (REPOBASS II).