3 Causes Cardiomyocyte Apoptosis and Cardiomyopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alopecia in a Viable Phospholipase C Delta 1 and Phospholipase C Delta 3 Double Mutant
Alopecia in a Viable Phospholipase C Delta 1 and Phospholipase C Delta 3 Double Mutant Fabian Runkel1¤, Maik Hintze1,2, Sebastian Griesing1,2, Marion Michels1, Birgit Blanck1, Kiyoko Fukami3, Jean-Louis Gue´net4, Thomas Franz1* 1 Anatomisches Institut, Universita¨t Bonn, Bonn, Germany, 2 Studiengang Molekulare Biomedizin, LIMES, Bonn, Germany, 3 Laboratory of Genome and Biosignal, Tokyo University of Pharmacy and Life Science, Hachioji-city, Tokyo, Japan, 4 De´partement de Biologie du De´veloppement, Institut Pasteur, Paris, France Abstract Background: Inositol 1,4,5trisphosphate (IP3) and diacylglycerol (DAG) are important intracellular signalling molecules in various tissues. They are generated by the phospholipase C family of enzymes, of which phospholipase C delta (PLCD) forms one class. Studies with functional inactivation of Plcd isozyme encoding genes in mice have revealed that loss of both Plcd1 and Plcd3 causes early embryonic death. Inactivation of Plcd1 alone causes loss of hair (alopecia), whereas inactivation of Plcd3 alone has no apparent phenotypic effect. To investigate a possible synergy of Plcd1 and Plcd3 in postnatal mice, novel mutations of these genes compatible with life after birth need to be found. Methodology/Principal Findings: We characterise a novel mouse mutant with a spontaneously arisen mutation in Plcd3 (Plcd3mNab) that resulted from the insertion of an intracisternal A particle (IAP) into intron 2 of the Plcd3 gene. This mutation leads to the predominant expression of a truncated PLCD3 protein lacking the N-terminal PH domain. C3H mice that carry one or two mutant Plcd3mNab alleles are phenotypically normal. However, the presence of one Plcd3mNab allele exacerbates the alopecia caused by the loss of functional Plcd1 in Del(9)olt1Pas mutant mice with respect to the number of hair follicles affected and the body region involved. -

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Mouse Plcd3 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Plcd3 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Plcd3 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Plcd3 gene (NCBI Reference Sequence: NM_152813 ; Ensembl: ENSMUSG00000020937 ) is located on Mouse chromosome 11. 15 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 15 (Transcript: ENSMUST00000103077). Exon 9~10 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Plcd3 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-133L3 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 9 starts from about 59.53% of the coding region. The knockout of Exon 9~10 will result in frameshift of the gene. The size of intron 8 for 5'-loxP site insertion: 1710 bp, and the size of intron 10 for 3'-loxP site insertion: 714 bp. The size of effective cKO region: ~945 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 8 9 10 11 15 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Plcd3 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions
International Journal of Molecular Sciences Review The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions Csaba Máthé *, Márta M-Hamvas, Csongor Freytag and Tamás Garda Department of Botany, Faculty of Science and Technology, University of Debrecen, H-4032 Debrecen, Hungary; [email protected] (M.M.-H.); [email protected] (C.F.); [email protected] (T.G.) * Correspondence: [email protected] Abstract: The protein phosphatase PP2A is essential for the control of integrated eukaryotic cell functioning. Several cellular and developmental events, e.g., plant growth regulator (PGR) mediated signaling pathways are regulated by reversible phosphorylation of vesicle traffic proteins. Reviewing present knowledge on the relevant role of PP2A is timely. We discuss three aspects: (1) PP2A regulates microtubule-mediated vesicle delivery during cell plate assembly. PP2A dephosphorylates members of the microtubule associated protein family MAP65, promoting their binding to microtubules. Regulation of phosphatase activity leads to changes in microtubule organization, which affects vesicle traffic towards cell plate and vesicle fusion to build the new cell wall between dividing cells. (2) PP2A-mediated inhibition of target of rapamycin complex (TORC) dependent signaling pathways contributes to autophagy and this has possible connections to the brassinosteroid signaling pathway. (3) Transcytosis of vesicles transporting PIN auxin efflux carriers. PP2A regulates vesicle localization and recycling of PINs related to GNOM (a GTP–GDP exchange factor) mediated pathways. The proper intracellular traffic of PINs is essential for auxin distribution in the plant body, thus in whole Citation: Máthé, C.; M-Hamvas, M.; plant development. -
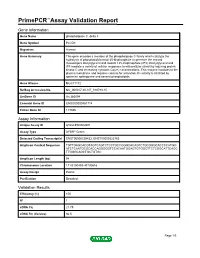
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name phospholipase C, delta 3 Gene Symbol PLCD3 Organism Human Gene Summary This gene encodes a member of the phospholipase C family which catalyze the hydrolysis of phosphatidylinositol 45-bisphosphate to generate the second messengers diacylglycerol and inositol 145-trisphosphate (IP3). Diacylglycerol and IP3 mediate a variety of cellular responses to extracellular stimuli by inducing protein kinase C and increasing cytosolic Ca(2+) concentrations. This enzyme localizes to the plasma membrane and requires calcium for activation. Its activity is inhibited by spermine sphingosine and several phospholipids. Gene Aliases MGC71172 RefSeq Accession No. NC_000017.10, NT_010783.15 UniGene ID Hs.380094 Ensembl Gene ID ENSG00000161714 Entrez Gene ID 113026 Assay Information Unique Assay ID qHsaCED0002601 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000539433, ENST00000322765 Amplicon Context Sequence TGTTGAGCACGTAGTCAGTCTCCTGCCGGGCACAGTCTGCGGGCACCCCATGG ATCTCAATGCGCACCAGGGGGTCCACAATGGAGTGTGGCTTCTCGGCATTCAGC TTGGGCAGCTGCTGTGC Amplicon Length (bp) 94 Chromosome Location 17:43190493-43190616 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 1 cDNA Cq 21.75 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 23.62 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLCD3, Human Amplification Plot Amplification of cDNA generated from -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Stimulation of Phospholipid Metabolism in Embryonic Muscle
Proc. Natl. Acad. Sci. USA Vol. 76, No. 9, pp. 4474-4478, September 1979 Cell Biology Stimulation of phospholipid metabolism in embryonic muscle cells treated with phospholipase C (phospholipid synthesis/myogenesis) CLAUDIA KENT Department of Biochemistry, Purdue University, West Lafayette, Indiana 47907 Communicated by Edwin T. Mertz, May 29, 1979 ABSTRACT Phospholipid metabolism is dramatically MATERIALS AND METHODS stimulated in cultured myogenic cells in which cell fusion was inhibited with phospholipase C (phosphatidylcholine choline- Cultured Cells. Pectoral muscle from 11-day chicken em- phosphohydrolase; EC 3.1.4.3). Phospholipase C was active bryos was dissected, loose connective tissue was removed, and under the culture conditions as shown by the degradation of the muscle was minced into 1- to 2-mm fragments. Cells were exogenous phosphatidylcholine. Rates of incorporation of 32p; dissociated from the tissue fragments by trituration with a and [metkyl-3Hlcholine into lipids were about 5-fold greater in phospholipase-treated cells than in either untreated fusing cells pasteur pipette (8) in calcium- and magnesium-free Earle's salt or untreated cells prevented from fusing by-calcium deprivation. solution. The cell suspension was filtered through cheesecloth, The greatest stimulation in the phospholipase C-treated cultures preplated for 15 min (9), and then diluted with culture medium occurred with synthesis of phospai tlcholine and sphin- to 5 X 105 cells/ml. The cells were plated in tissue culture dishes gomyelin; synthesis of phosphatidyinositol and cardiolipin was precoated with rat tail collagen (10) at 8 ml of cell suspension not stimulated. Degradation of cellular [32Plphosphatidylcholine and appearance in the culture medium of the degradation per 100 mm dish. -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -

Emerging Roles of PHLPP Phosphatases in Cell Signaling
PA61CH26_Newton ARjats.cls September 22, 2020 12:5 Annual Review of Pharmacology and Toxicology PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling Timothy R. Baffi,∗ Ksenya Cohen Katsenelson,∗ and Alexandra C. Newton Department of Pharmacology, University of California, San Diego, La Jolla, California 92093-0721, USA; email: [email protected] Annu. Rev. Pharmacol. Toxicol.2021. 61:26.1–26.21 Keywords The Annual Review of Pharmacology and Toxicology is PHLPP, Akt, PKC, phosphatase, phosphorylation, cancer, transcription online at pharmtox.annualreviews.org https://doi.org/10.1146/annurev-pharmtox-031820- Abstract 122108 Whereas protein kinases have been successfully targeted for a variety of dis- Copyright © 2021 by Annual Reviews. eases, protein phosphatases remain an underutilized therapeutic target, in All rights reserved part because of incomplete characterization of their effects on signaling net- ∗ These authors contributed equally to this article works. The pleckstrin homology domain leucine-rich repeat protein phos- Annu. Rev. Pharmacol. Toxicol. 2021.61. Downloaded from www.annualreviews.org phatase (PHLPP) is a relatively new player in the cell signaling field, and new Access provided by University of California - San Diego on 11/11/20. For personal use only. roles in controlling the balance among cell survival, proliferation, and apop- tosis are being increasingly identified. Originally characterized for its tumor- suppressive function in deactivating the prosurvival kinase Akt, PHLPP may have an opposing role in promoting survival, as recent evidence suggests. Additionally, identification of the transcription factor STAT1 as a substrate unveils a role for PHLPP as a critical mediator of transcriptional programs in cancer and the inflammatory response. -

Mouse ENPP7 / NPP-7 Protein (His Tag)
Mouse ENPP7 / NPP-7 Protein (His Tag) Catalog Number: 50567-M08H General Information SDS-PAGE: Gene Name Synonym: ALK-SMase; E-NPP7; NPP-7; NPP7; Alk-SMase; Gm254 Protein Construction: A DNA sequence encoding the mouse ENPP7 (Q3TIW9) (Met 1-Gln 421) was expressed, with a polyhistidine tag at the C-terminus. Source: Mouse Expression Host: Human Cells QC Testing Purity: > 90 % as determined by SDS-PAGE Endotoxin: Protein Description < 1.0 EU per μg of the protein as determined by the LAL method Mouse Ectonucleotide pyrophosphatase / phosphodiesterase family Stability: member 7, also known as Alkaline sphingomyelin phosphodiesterase, Intestinal alkaline sphingomyelinase, Alk-Smase, ENPP7 and NPP-7, is a Samples are stable for up to twelve months from date of receipt at -70 ℃ single-pass type I membrane protein which belongs to the nucleotide pyrophosphatase / phosphodiesterase family. ENPP7 / NPP-7 is Predicted N terminal: Ala 22 expressed in the intestines and human bile. ENPP7 / NPP-7 is localized at Molecular Mass: the surface of the microvillar membrane in small intestine enterocytes, as well as in endosome-like structures and in Golgi complex. The main The secreted recombinant mouse ENPP7 consists of 411 amino acids and function of ENPP7 / NPP-7 is to convert the dietary sphingomyelin into has a calculated molecular mass of 47 kDa. It migrates as an ceramide, the sphingolipid messengers via hydrolyzation. ENPP7 / NPP-7 approximately 60 kDa band in SDS-PAGE under reducing conditions due is also reported to exert a phospholipase C activity toward palmitoyl lyso- to glycosylation. phosphocholine. The activity of this enzyme is inhibited in a dose dependent manner by ATP, imidazole, orthovanadate and zinc ion. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in