A Molecular Cytogenetic Investigation of Secondary Abnormalities and Clonal Evolution in ETV6-RUNX1 Positive Acute Lymphoblastic Leukaemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Compensation Among HMGN Variants Modulates the Dnase I Hypersensitive Sites at Enhancers
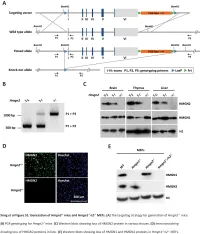
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press Research Functional compensation among HMGN variants modulates the DNase I hypersensitive sites at enhancers Tao Deng,1,12 Z. Iris Zhu,2,12 Shaofei Zhang,1 Yuri Postnikov,1 Di Huang,2 Marion Horsch,3 Takashi Furusawa,1 Johannes Beckers,3,4,5 Jan Rozman,3,5 Martin Klingenspor,6,7 Oana Amarie,3,8 Jochen Graw,3,8 Birgit Rathkolb,3,5,9 Eckhard Wolf,9 Thure Adler,3 Dirk H. Busch,10 Valérie Gailus-Durner,3 Helmut Fuchs,3 Martin Hrabeˇ de Angelis,3,4,5 Arjan van der Velde,2,13 Lino Tessarollo,11 Ivan Ovcherenko,2 David Landsman,2 and Michael Bustin1 1Protein Section, Laboratory of Metabolism, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA; 2Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland 20892, USA; 3German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 4Experimental Genetics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, 85354 Freising-Weihenstephan, Germany; 5German Center for Diabetes Research (DZD), 85764 Neuherberg, Germany; 6Molecular Nutritional Medicine, Technische Universität München, 85350 Freising, Germany; 7Center for Nutrition and Food Sciences, Technische Universität München, 85350 Freising, Germany; 8Institute of Developmental Genetics (IDG), 85764 -

Genetic Variants in Humanin Nuclear Isoform Gene Regions Show No Association with Coronary Artery Disease
Genetic variants in humanin nuclear isoform gene regions show no association with coronary artery disease Mall Eltermaa ( [email protected] ) University of Tartu https://orcid.org/0000-0002-9651-4107 Maili Jakobson University of Tartu Meeme Utt University of Tartu Sulev Kõks Perron Institute for Neurological and Translational Science Reedik Mägi University of Tartu Joel Starkopf Tartu University Hospital Research article Keywords: humanin, humanin-like, coronary artery disease, association study, peptide Posted Date: June 25th, 2019 DOI: https://doi.org/10.21203/rs.2.10582/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Research Notes on November 21st, 2019. See the published version at https://doi.org/10.1186/s13104-019-4807-x. Loading [MathJax]/jax/output/CommonHTML/jax.js Page 1/7 Abstract Background Coronary artery disease contributes to noncommunicable disease deaths worldwide. In order to make preventive methods more accurate, we need to know more about the development and progress of this pathology, including the genetic aspects. Humanin is a small peptide known for its cytoprotective and anti-apoptotic properties. Our study looked for genomic associations between humanin-like nuclear isoform genes and coronary artery disease using CARDIoGRAMplusC4D Consortium data. Results Lookup from meta-analysis datasets gave single nucleotide polymorphisms in all 13 humanin-like nuclear isoform genes with the lowest P-value for rs6151662 from the MTRNR2L2 gene including the 50 kb anking region in both directions (P-value = 0.0037). Within the gene region alone the top variant was rs78083998 from the MTRNR2L13 region (meta-analysis P-value=0.042). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

A Clinicopathological and Molecular Genetic Analysis of Low-Grade Glioma in Adults
A CLINICOPATHOLOGICAL AND MOLECULAR GENETIC ANALYSIS OF LOW-GRADE GLIOMA IN ADULTS Presented by ANUSHREE SINGH MSc A thesis submitted in partial fulfilment of the requirements of the University of Wolverhampton for the degree of Doctor of Philosophy Brain Tumour Research Centre Research Institute in Healthcare Sciences Faculty of Science and Engineering University of Wolverhampton November 2014 i DECLARATION This work or any part thereof has not previously been presented in any form to the University or to any other body whether for the purposes of assessment, publication or for any other purpose (unless otherwise indicated). Save for any express acknowledgments, references and/or bibliographies cited in the work, I confirm that the intellectual content of the work is the result of my own efforts and of no other person. The right of Anushree Singh to be identified as author of this work is asserted in accordance with ss.77 and 78 of the Copyright, Designs and Patents Act 1988. At this date copyright is owned by the author. Signature: Anushree Date: 30th November 2014 ii ABSTRACT The aim of the study was to identify molecular markers that can determine progression of low grade glioma. This was done using various approaches such as IDH1 and IDH2 mutation analysis, MGMT methylation analysis, copy number analysis using array comparative genomic hybridisation and identification of differentially expressed miRNAs using miRNA microarray analysis. IDH1 mutation was present at a frequency of 71% in low grade glioma and was identified as an independent marker for improved OS in a multivariate analysis, which confirms the previous findings in low grade glioma studies. -

Functional Annotations of Single-Nucleotide Polymorphism
CLINICAL RESEARCH e-ISSN 1643-3750 © Med Sci Monit, 2020; 26: e922710 DOI: 10.12659/MSM.922710 Received: 2020.01.08 Accepted: 2020.02.20 Functional Annotations of Single-Nucleotide Available online: 2020.03.30 Published: 2020.05.25 Polymorphism (SNP)-Based and Gene-Based Genome-Wide Association Studies Show Genes Affecting Keratitis Susceptibility Authors’ Contribution: BCDEF 1 Yue Xu* 1 Department of Ophthalmology, First Affiliated Hospital of Soochow University, Study Design A BCDEF 2 Xiao-Lin Yang* Suzhou, Jiangsu, P.R. China Data Collection B 2 Center for Genetic Epidemiology and Genomics, School of Public Health, Medical Statistical Analysis C BCD 1 Xiao-Long Yang College of Soochow University, Suzhou, Jiangsu, P.R. China Data Interpretation D BC 1 Ya-Ru Ren Manuscript Preparation E BC 1 Xin-Yu Zhuang Literature Search F Funds Collection G ADE 2 Lei Zhang ADE 1 Xiao-Feng Zhang * Yue Xu and Xiao-Lin Yang contributed equally Corresponding Authors: Xiao-Feng Zhang, e-mail: [email protected], Lei Zhang, e-mail: [email protected] Source of support: Departmental sources Background: Keratitis is a complex condition in humans and is the second most common cause of legal blindness worldwide. Material/Methods: To reveal the genomic loci underlying keratitis, we performed functional annotations of SNP-based and gene- based genome-wide association studies of keratitis in the UK Biobank (UKB) cohort with 337 199 subjects of European ancestry. Results: The publicly available SNP-based association results showed a total of 34 SNPs, from 14 distinct loci, associated with keratitis in the UKB. Gene-based association analysis identified 2 significant genes:IQCF3 (p=2.0×10–6) and SOD3 (p=2.0×10–6). -

Molecular Characterization of Acute Myeloid Leukemia by Next Generation Sequencing: Identification of Novel Biomarkers and Targets of Personalized Therapies
Alma Mater Studiorum – Università di Bologna Dipartimento di Medicina Specialistica, Diagnostica e Sperimentale Dottorato di Ricerca in Oncologia, Ematologia e Patologia XXX Ciclo Settore Scientifico Disciplinare: MED/15 Settore Concorsuale:06/D3 Molecular characterization of acute myeloid leukemia by Next Generation Sequencing: identification of novel biomarkers and targets of personalized therapies Presentata da: Antonella Padella Coordinatore Prof. Pier-Luigi Lollini Supervisore: Prof. Giovanni Martinelli Esame finale anno 2018 Abstract Acute myeloid leukemia (AML) is a hematopoietic neoplasm that affects myeloid progenitor cells and it is one of the malignancies best studied by next generation sequencing (NGS), showing a highly heterogeneous genetic background. The aim of the study was to characterize the molecular landscape of 2 subgroups of AML patients carrying either chromosomal number alterations (i.e. aneuploidy) or rare fusion genes. We performed whole exome sequencing and we integrated the mutational data with transcriptomic and copy number analysis. We identified the cell cycle, the protein degradation, response to reactive oxygen species, energy metabolism and biosynthetic process as the pathways mostly targeted by alterations in aneuploid AML. Moreover, we identified a 3-gene expression signature including RAD50, PLK1 and CDC20 that characterize this subgroup. Taking advantage of RNA sequencing we aimed at the discovery of novel and rare gene fusions. We detected 9 rare chimeric transcripts, of which partner genes were transcription factors (ZEB2, BCL11B and MAFK) or tumor suppressors (SAV1 and PUF60) rarely translocated across cancer types. Moreover, we detected cryptic events hiding the loss of NF1 and WT1, two recurrently altered genes in AML. Finally, we explored the oncogenic potential of the ZEB2-BCL11B fusion, which revealed no transforming ability in vitro. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

HMGB1 in Health and Disease R
Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2014 HMGB1 in health and disease R. Kang R. C. Chen Q. H. Zhang W. Hou S. Wu See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Emergency Medicine Commons Recommended Citation Kang R, Chen R, Zhang Q, Hou W, Wu S, Fan X, Yan Z, Sun X, Wang H, Tang D, . HMGB1 in health and disease. 2014 Jan 01; 40():Article 533 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/533. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors R. Kang, R. C. Chen, Q. H. Zhang, W. Hou, S. Wu, X. G. Fan, Z. W. Yan, X. F. Sun, H. C. Wang, D. L. Tang, and +8 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/533 NIH Public Access Author Manuscript Mol Aspects Med. Author manuscript; available in PMC 2015 December 01. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Aspects Med. 2014 December ; 0: 1–116. doi:10.1016/j.mam.2014.05.001. HMGB1 in Health and Disease Rui Kang1,*, Ruochan Chen1, Qiuhong Zhang1, Wen Hou1, Sha Wu1, Lizhi Cao2, Jin Huang3, Yan Yu2, Xue-gong Fan4, Zhengwen Yan1,5, Xiaofang Sun6, Haichao Wang7, Qingde Wang1, Allan Tsung1, Timothy R. -

CSE642 Final Version
Eindhoven University of Technology MASTER Dimensionality reduction of gene expression data Arts, S. Award date: 2018 Link to publication Disclaimer This document contains a student thesis (bachelor's or master's), as authored by a student at Eindhoven University of Technology. Student theses are made available in the TU/e repository upon obtaining the required degree. The grade received is not published on the document as presented in the repository. The required complexity or quality of research of student theses may vary by program, and the required minimum study period may vary in duration. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain Eindhoven University of Technology MASTER THESIS Dimensionality Reduction of Gene Expression Data Author: S. (Sako) Arts Daily Supervisor: dr. V. (Vlado) Menkovski Graduation Committee: dr. V. (Vlado) Menkovski dr. D.C. (Decebal) Mocanu dr. N. (Nikolay) Yakovets May 16, 2018 v1.0 Abstract The focus of this thesis is dimensionality reduction of gene expression data. I propose and test a framework that deploys linear prediction algorithms resulting in a reduced set of selected genes relevant to a specified case. Abstract In cancer research there is a large need to automate parts of the process of diagnosis, this is mainly to reduce cost, make it faster and more accurate. -
Supplemental Data.Pdf
Table S1. Summary of sequencing results A. DNase‐seq data % Align % Mismatch % >=Q30 bases Sample ID # Reads % unique reads (PF) Rate (PF) (PF) WT_1 84,301,522 86.76 0.52 93.34 96.30 WT_2 98,744,222 84.97 0.51 93.75 89.94 Hmgn1‐/‐_1 79,620,656 83.94 0.86 90.58 88.65 Hmgn1‐/‐_2 62,673,782 84.13 0.87 91.30 89.18 Hmgn2‐/‐_1 87,734,440 83.49 0.71 91.81 90.00 Hmgn2‐/‐_2 82,498,808 83.25 0.69 92.73 90.66 Hmgn1‐/‐n2‐/‐_1 71,739,638 68.51 2.31 81.11 89.22 Hmgn1‐/‐n2‐/‐_2 74,113,682 68.19 2.37 81.16 86.57 B. ChIP‐seq data Histone % Align % Mismatch % >=Q30 % unique Genotypes # Reads marks (PF) Rate (PF) bases (PF) reads H3K4me1 100670054 92.99 0.28 91.21 87.29 H3K4me3 67064272 91.97 0.35 89.11 27.15 WT H3K27ac 90,340,242 93.57 0.28 95.02 89.80 input 111,292,572 78.24 0.55 96.07 86.99 H3K4me1 84598176 92.34 0.33 91.2 81.69 H3K4me3 90032064 92.19 0.44 88.76 15.81 Hmgn1‐/‐n2‐/‐ H3K27ac 86,260,526 93.40 0.29 94.94 87.49 input 78,142,334 78.47 0.56 95.82 81.11 C. MNase‐seq data % Mismatch % >=Q30 bases % unique Sample ID # Reads % Align (PF) Rate (PF) (PF) reads WT_1_Extensive 45,232,694 55.23 1.49 90.22 81.73 WT_1_Limited 105,460,950 58.03 1.39 90.81 79.62 WT_2_Extensive 40,785,338 67.34 1.06 89.76 89.60 WT_2_Limited 105,738,078 68.34 1.05 90.29 85.96 Hmgn1‐/‐n2‐/‐_1_Extensive 117,927,050 55.74 1.49 89.50 78.01 Hmgn1‐/‐n2‐/‐_1_Limited 61,846,742 63.76 1.22 90.57 84.55 Hmgn1‐/‐n2‐/‐_2_Extensive 137,673,830 60.04 1.30 89.28 78.99 Hmgn1‐/‐n2‐/‐_2_Limited 45,696,614 62.70 1.21 90.71 85.52 D. -

Characterisation of the Genomic Landscape of CRLF2‐Rearranged Acute Lymphoblastic Leukemia
Characterisation of the Genomic Landscape of CRLF2- rearranged Acute Lymphoblastic Leukemia Lisa J Russell1*, Lisa Jones1, Amir Enshaei1, Stefano Tonin1, Sarra L Ryan1, Jeyanthy Eswaran1 , Sirintra Nakjang2, Elli Papaemmanuil3,4, Jose M C Tubio4, Adele K Fielding5, Ajay Vora6, Peter J Campbell4, Anthony V Moorman1, and Christine J Harrison1 1 Leukaemia Research Cytogenetics Group, Northern Institute for Cancer Research, Newcastle University, Newcastle-upon-Tyne, UK 2 Bioinformatics Support Unit, Newcastle University, Newcastle-upon-Tyne, UK 3 Memorial Sloan Kettering Cancer Center, USA 4 Cancer Genome Project, Wellcome Trust Sanger Institute, Hinxton, UK 5 Research Department of Haemaoloty, UCL Cancer Institute, London, UK 6 Department of Haematology, Sheffield Children’s Hospital, Sheffield, UK; AVM and CJH contributed equally to this study Running Title – Genomic landscape of CRLF2 rearranged leukemia Correspondence to: Dr Lisa J Russell, Wolfson Childhood Cancer Research Centre, Northern Institute for Cancer Research, Newcastle University, Level 6, Herschel Building, Brewery Lane, Newcastle upon Tyne, NE1 7RU, [email protected]. Acknowledgements Support by: The Kay Kendall Leukaemia Fund, Leuka, European Haematology Association and Bloodwise (formerly Leukaemia and Lymphoma Research) This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as an ‘Accepted Article’, doi: 10.1002/gcc.22439 This article is protected by copyright. All rights reserved. Genes, Chromosomes & Cancer Page 2 of 147 Deregulated expression of the type I cytokine receptor, CRLF2, is observed in 5-15% of precursor B-cell acute lymphoblastic leukaemia (B-ALL).