STUDIES RELATED to the SYNTHESIS of a Thesis Presented
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
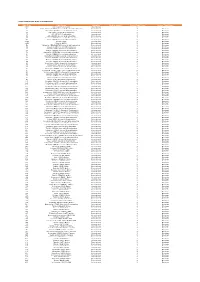
Codes Used in D&M
CODES USED IN D&M - MCPS A DISTRIBUTIONS D&M Code D&M Name Category Further details Source Type Code Source Type Name Z98 UK/Ireland Commercial International 2 20 South African (SAMRO) General & Broadcasting (TV only) International 3 Overseas 21 Australian (APRA) General & Broadcasting International 3 Overseas 36 USA (BMI) General & Broadcasting International 3 Overseas 38 USA (SESAC) Broadcasting International 3 Overseas 39 USA (ASCAP) General & Broadcasting International 3 Overseas 47 Japanese (JASRAC) General & Broadcasting International 3 Overseas 48 Israeli (ACUM) General & Broadcasting International 3 Overseas 048M Norway (NCB) International 3 Overseas 049M Algeria (ONDA) International 3 Overseas 58 Bulgarian (MUSICAUTOR) General & Broadcasting International 3 Overseas 62 Russian (RAO) General & Broadcasting International 3 Overseas 74 Austrian (AKM) General & Broadcasting International 3 Overseas 75 Belgian (SABAM) General & Broadcasting International 3 Overseas 79 Hungarian (ARTISJUS) General & Broadcasting International 3 Overseas 80 Danish (KODA) General & Broadcasting International 3 Overseas 81 Netherlands (BUMA) General & Broadcasting International 3 Overseas 83 Finnish (TEOSTO) General & Broadcasting International 3 Overseas 84 French (SACEM) General & Broadcasting International 3 Overseas 85 German (GEMA) General & Broadcasting International 3 Overseas 86 Hong Kong (CASH) General & Broadcasting International 3 Overseas 87 Italian (SIAE) General & Broadcasting International 3 Overseas 88 Mexican (SACM) General & Broadcasting -

Handbook Courage
MEDGAR EVERS COLLEGE, CUNY STUDENT 2014 - 2015 HANDBOOK COURAGE. STRENGTH. FORTITUDE. 2014 - 2015 1 COURAGE. STRENGTH. FORTITUDE. Dear Student, It gives me great pleasure to welcome you to Medgar Evers College (MEC). Congratulations on your decision to enroll at this great institution. This is the beginning of an important milestone in your career. The founders’ vision for the College resonates today as MEC learning for students in Central Brooklyn and beyond. Our stellar programs, student support services, fulfills its and mandate dedicated to be faculty a world-renowned and staff are the institution foundations of higher on which you will build a successful academic and professional career. As your President, I am willing and ready to serve you unreservedly. I am committed to creating and fostering a learning environment that involves thoughtfulness, high expectations and a strong connection to the community. Moreover, I will ensure you receive a quality education – one that Aswill a enableMEC student, you to succeedI urge you in toan make ever-changing the most ofglobal the manyeconomy. opportunities we offer, both inside and outside of the classroom. Seek advisement, ask questions, attend classes, be on time, and curricular activities. From career preparation to cultural enrichment, the programs and facilities availableparticipate on in campus discussions. can help Get you involved acquire in ourmany student valuable clubs, skills organizations and experiences. and other extra- I urge you to explore this handbook thoroughly as it contains essential information about the College’s policies, procedures, resources and campus services. Make it your guide through your time here at Medgar Evers College. -
Citations of Articles by Some Nobel Prize Winners, J.Informetrics 9(2015)466
Всички цитати Звено: ( ИФХ ) Институт по физикохимия „Академик Ростислав Каишев” Година: 2015 ÷ 2015 Брой цитирани публикации: 576 Брой цитиращи източници: 1762 1959 1. Scheludko, A., Exerowa, D., Uber den elektrostatischen Druck in Schaumfilmen aus wasserigen Elektrolytlosungen. Kolloid-Zeitschrift, 165, 2, Springer-Verlag, 1959, ISSN:0303402X, DOI:10.1007/BF01809974, 148 - 151 Цитира се в: 1. Danov, K.D., Kralchevsky, P.A., Radulova, G.M., Basheva, E.S., Stoyanov, S.D., Pelan, E.G., Shear rheology of mixed protein adsorption layers vs their structure studied by surface force measurements, Advances in Colloid and Interface Science 222, 148-161 (2015) 1960 2. Scheludko, A., Exerowa, D., Uber den elektrostatischen und van der Waalsschen zusatzlichen Druck in wasserigen Schaumfilmen. Kolloid-Zeitschrift, 168, 1, Springer-Verlag, 1960, ISSN:0303402X, DOI:10.1007/BF01513550, 24 - 28 Цитира се в: 2. Dhayal, S.K., Delahaije, R.J.B.M., De Vries, R.J., Gruppen, H., Wierenga, P.A., Enzymatic cross-linking of α-lactalbumin to produce nanoparticles with increased foam stability, Soft Matter, 11 (40), pp. 7888-7898 (2015) 3. Peng, T., Peng, K., Li, Q., Methodology for disjoining pressure of free water nanofilms, Journal of Physical Chemistry C, 119 (25), pp. 14273-14280 (2015) 1969 3. Kashchiev, D., Nucleation at variable supersaturation. Surface Science, 18, 2, 1969, ISSN:396028, 293 - 297 Цитира се в: 4. M.A.Duran-Olivencia, J.F.Lutsko, Mesoscopic nucleation theory for confined systems: A one- parameter model, Phys.Rev. E 91(2015)022402. 5. MA Durán-Olivencia, JF Lutsko, Unification of classical nucleation theories via unified Ito- Stratonovich stochastic equation, Phys.Rev. -

United States Patent (10) Patent No.: US 7,379,721 B2 Doudnikoff Et Al
US007379721B2 (12) United States Patent (10) Patent No.: US 7,379,721 B2 Doudnikoff et al. (45) Date of Patent: May 27, 2008 (54) RADIO STATION PREFERENCE 6,374,177 B1 * 4/2002 Lee et al. ................... TO1,200 TRANSLATIONAL MAPPING 6,445,306 B1* 9/2002 Trovato et al. ........ 340/825.24 6,526,411 B1 2/2003 Ward .......................... 707/102 (75) Inventors: Gregory Michael Doudnikoff, Raleigh, 96.36W w R. 2. SRIllillale et . al.. ........... .4.s S. (US) past. y Esili 7,171,174 B2 * 1/2007 Ellis et al. ............ ... 455,132 1nes, ; Steven Vicnae VIIIer, 7.251,452 B2 * 7/2007 Stumphauzer, II ......... 455/3.06 Cary, NC (US) 2002/0142722 A1* 10, 2002 Gutta et al. ................... 455/45 2003, OO13425 A1* 1/2003 ... 455,186.1 (73) Assignee: International Business Machines 2003/0083028 A1* 5/2003 Williamson .............. 455,186.1 Corporation, Armonk, NY (US) * cited by examiner (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 Primary Examiner Duc M Nguyen U.S.C. 154(b) by 443 days. (74) Attorney, Agent, or Firm—VanLeeuwen & VanLeeuwen: Andre M. Gibbs (21) Appl. No.: 11/190,401 (57) ABSTRACT (22) Filed: Jul. 27, 2005 A user's media preferences can be obtained and stored for (65) Prior Publication Data later use in setting up a currently-available media device. The media device inputs are programmed based on the US 2007/OO37534 A1 Feb. 15, 2007 media device's capabilities and the available media sources (51) Int. Cl. forOr the particularpart1Cular geographigeograph1C area 1nin Whichwhich ththe uSer 1S H04B I/8 (2006.01) currently located. -

Fisher Communications, Inc
D S Fisher Communications, Inc. > $ sa in w ga lo d lo w Th th fa > re co Fisher Annual a st w ic do Report 02 try p an p le an na pe sp N W I a co CORP: DOCUMENT: YEAR: sp of Fisher fo Communications, Annual Report / 10-K 2002 b Inc. an ac DOC Fi Dear Shareholders Hopes for a profitable 2002 did not the cornerstones of our restructuring program: > materialize. Consolidated net loss was concentrating on our core businesses; increasing $.23 a share in 2002. This includes gains on the revenue base; streamlining operations and sale of real estate and net gain on derivative controlling expenses; and selling selected assets to instruments. If, for purposes of comparison pay down debt. with 2001 results, the real estate and derivative gains, as well as a charge for expensing deferred Concentrating on Core Businesses In Fisher’s loan costs in connection with refinancing of 1999 annual report I stated that “Fisher is focusing debt, were excluded from 2002 results, the net its efforts and resources on becoming a fully loss per share would have been $1.35, compared integrated communications and media company.” with a $.96 per share loss reported in 2001. So began a series of actions involving the acquisition of 11 television stations in 1999, sale of Fisher’s The broad context for Fisher’s performance and legacy flour milling businesses in 2001, construction that of many other Northwest companies is a of Fisher Plaza, and the sale of commercial real faltering national economy and an even weaker estate and other selected assets in 2002 and 2003. -

Notre Dame Athletics
NOTRE DAME 2008-09 MEN’S BASKETBALL 2008-09 Schedule OCTOBER 31 (9/9) BRIAR CLIFF (EXH) W, 103-64 NOVEMBER 9 (9/9) STONEHILL COLLEGE (EXH) W, 79-47 16 (9/9) USC UPSTATE W, 94-58 21 (8/9) at Loyola Marymount W, 65-54 EA Sports Maui Invitational Lahaina, Maui, Hawaii 24 (8/8) vs. Indiana (ESPN2) W, 88-50 25 (8/8) vs. Texas (6/7) (ESPN) W, 81-80 March 10-14, 2009 26 (8/8) vs. North Carolina (1/1) (ESPN) L, 87-102 Madison Square Garden (19,786) • New York, N.Y. 30 (8/8) FURMAN W, 93-61 TV: ESPN DECEMBER Sean McDonough (play-by-play analyst) 2 (7/7) SOUTH DAKOTA W, 102-76 Bill Raftery and Jay Bilas (color analysts) The Hartford Hall of Fame Showcase Allen Hopkins (sideline reporter) Indianapolis, Ind. 6 (7/7) vs. Ohio State (ESPNU) L, 62-67 Radio: Jack Nolan (play-by-play analyst) 13 (12/13) BOSTON UNIVERSITY W, 74-67 LaPhonso Ellis (color analyst) 20 (12/14) DELAWARE STATE W, 88-50 Notre Dame Sports Properties originates the Notre Dame Radio Net- 22 (12/14) SAVANNAH STATE W, 81-49 31 (7/10) at DePaul* (ESPN2) W, 92-82 work which includes: WLS 890 AM in Chicago, Ill (Chicagoland area and JANUARY Midwest); WSBT 960 AM in South Bend, Ind.; XL950 AM in Indianapo- 3 (7/10) at St. John’s* (ESPNU) L, 65-71 lis, Ind.; WEFM95.9 FM in Michigan City and Gary, Ind.; WKKX 1600 AM 5 (13/13) GEORGETOWN* (9/10) (ESPN) W, 73-67 in Wheeling, W. -

Germania Sacra Die Kirche Des Alten Reiches Und Ihre Institutionen
GERMANIA SACRA DIE KIRCHE DES ALTEN REICHES UND IHRE INSTITUTIONEN HERAUSGEGEBEN VON DER AKADEMIE DER WISSENSCHAFTEN ZU GÖTTINGEN UNTER DER LEITUNG VON HEDWIG RÖCKELEIN REDAKTION JASMIN HOVEN-HACKER · BÄRBEL KRÖGER NATHALIE KRUPPA · CHRISTIAN POPP AKADEMIE DER WISSENSCHAFTEN ZU GÖTTINGEN Steinfeld_Joester_PersonalS2,2.indb 1 21.08.2018 15:42:47 Steinfeld_Joester_PersonalS2,2.indb 2 21.08.2018 15:42:47 SUPPLEMENTBAND 2,2 ÄBTE UND CHORHERREN DES PRÄMONSTRATENSERSTIFTS STEINFELD BEARBEITET VON INGRID JOESTER AKADEMIE DER WISSENSCHAFTEN ZU GÖTTINGEN Steinfeld_Joester_PersonalS2,2.indb 3 21.08.2018 15:42:47 Dieser Band wurde durch die Gemeinsame Wissenschaftskonferenz (GWK) im Rahmen des Akademienprogramms mit Mitteln des Bundes und des Landes Niedersachsen gefördert. ISBN 978-3-946048-15-2 Diese Publikation ist elektronisch verfügbar auf dem Dokumentenserver der Akademie der Wissenschaften zu Göttingen URI: http://hdl.handle.net/11858/00-001S-0000-002D-B56E-3 Bibliografische Information der Deutschen Nationalbibliothek Die Deutsche Nationalbibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie; detaillierte bibliografische Daten sind im Internet über http://dnb.dnb.de abrufbar. © 2018 Akademie der Wissenschaften zu Göttingen Steinfeld_Joester_PersonalS2,2.indb 4 21.08.2018 15:42:47 INHALTSVERZEICHNIS TEILBAND 1 Vorwort . V Siglen und Abkürzungen . IX 1 . Quellen und Literatur . 1 a . Ungedruckte Quellen . 1 b . Gedruckte Quellen . 10 c . Literatur . 16 d . Bei der Bearbeitung der Lebensläufe der Pröpste und Äbte berücksichtigte Chroniken . 28 e . Bei der Bearbeitung der Chorherrenliste berücksichtigte Quellen bzw . Literatur . 33 2 . Ämter . 39 a . Prioren . 39 b . Subprioren . 42 c . Zirkatoren . 44 d . Novizenmeister . 46 e . Provisor, Ce(l)l(er)arius, Vestiarius . 49 f . Kantoren, Succentoren . 51 g .