Open Research Online Oro.Open.Ac.Uk
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Peking University-Juntendo University Joint Symposium on Cancer Research and Treatment ADAM28 (A Disintegrin and Metalloproteinase 28) in Cancer Cell Proliferation and Progression
Whatʼs New from Juntendo University, Tokyo Juntendo Medical Journal 2017. 63(5), 322-325 Peking University - Juntendo University Joint Symposium on Cancer Research and Treatment ADAM28 (a Disintegrin and Metalloproteinase 28) in Cancer Cell Proliferation and Progression YASUNORI OKADA* *Department of Pathophysiology for Locomotive and Neoplastic Diseases, Juntendo University Graduate School of Medicine, Tokyo, Japan A disintegrinandmetalloproteinase 28 (ADAM28) is overexpressedpredominantlyby carcinoma cells in more than 70% of the non-small cell lung carcinomas, showing positive correlations with carcinoma cell proliferation and metastasis. ADAM28 cleaves insulin-like growth factor binding protein-3 (IGFBP-3) in the IGF-I/IGFBP-3 complex, leading to stimulation of cell proliferation by intact IGF-I released from the complex. ADAM28 also degrades von Willebrand factor (VWF), which induces apoptosis in human carcinoma cell lines with negligible ADAM28 expression, andthe VWF digestionby ADAM28-expressing carcinoma cells facilitates them to escape from VWF-induced apoptosis, resulting in promotion of metastasis. We have developed human antibodies against ADAM28 andshown that one of them significantly inhibits tumor growth andmetastasis using lung adenocarcinoma cells. Our data suggest that ADAM28 may be a new molecular target for therapy of the patients with ADAM28-expressing non-small cell lung carcinoma. Key words: a disintegrin and metalloproteinase 28 (ADAM28), cell proliferation, invasion, metastasis, human antibody inhibitor Introduction human cancers 2). However, development of the synthetic inhibitors of MMPs andtheir application Cancer cell proliferation andprogression are for treatment of the cancer patients failed 3). modulated by proteolytic cleavage of tissue micro- On the other hand, members of the ADAM (a environmental factors such as extracellular matrix disintegrin and metalloproteinase) gene family, (ECM), growth factors andcytokines, receptors another family belonging to the metzincin gene andcell adhesionmolecules. -

Conservation and Divergence of ADAM Family Proteins in the Xenopus Genome
Wei et al. BMC Evolutionary Biology 2010, 10:211 http://www.biomedcentral.com/1471-2148/10/211 RESEARCH ARTICLE Open Access ConservationResearch article and divergence of ADAM family proteins in the Xenopus genome Shuo Wei*1, Charles A Whittaker2, Guofeng Xu1, Lance C Bridges1,3, Anoop Shah1, Judith M White1 and Douglas W DeSimone1 Abstract Background: Members of the disintegrin metalloproteinase (ADAM) family play important roles in cellular and developmental processes through their functions as proteases and/or binding partners for other proteins. The amphibian Xenopus has long been used as a model for early vertebrate development, but genome-wide analyses for large gene families were not possible until the recent completion of the X. tropicalis genome sequence and the availability of large scale expression sequence tag (EST) databases. In this study we carried out a systematic analysis of the X. tropicalis genome and uncovered several interesting features of ADAM genes in this species. Results: Based on the X. tropicalis genome sequence and EST databases, we identified Xenopus orthologues of mammalian ADAMs and obtained full-length cDNA clones for these genes. The deduced protein sequences, synteny and exon-intron boundaries are conserved between most human and X. tropicalis orthologues. The alternative splicing patterns of certain Xenopus ADAM genes, such as adams 22 and 28, are similar to those of their mammalian orthologues. However, we were unable to identify an orthologue for ADAM7 or 8. The Xenopus orthologue of ADAM15, an active metalloproteinase in mammals, does not contain the conserved zinc-binding motif and is hence considered proteolytically inactive. We also found evidence for gain of ADAM genes in Xenopus as compared to other species. -
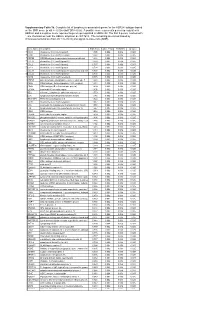
* Supplementary Table 3B. Complete List of Lymphocytic-Associated Genes
Supplementary Table 3b. Complete list of lymphocytic-associated genes for the HER2+I subtype based on the SNR score (p-val <= 0.005 and FDR<=0.05). A positive score represents genes up-regulated in HER2+I and a negative score represents genes up-regulated in HER2+NI. The first 9 genes, marked with *, are chemokines near the HER2+ amplicon at chr17q12. The remaining are sorted based by chromosomal location (from chr 1 to chr X) and signal-to-noise ratio (SNR). Gene Name Description SNR Score Feature P value FDR(BH) Q Value * CCL5 chemokine (C-C motif) ligand 5 1.395 0.002 0.036 0.025 * CCR7 chemokine (C-C motif) receptor 7 1.362 0.002 0.036 0.025 * CD79B CD79B antigen (immunoglobulin-associated beta) 1.248 0.002 0.036 0.025 * CCL13 chemokine (C-C motif) ligand 13 1.003 0.002 0.036 0.025 * CCL2 chemokine (C-C motif) ligand 2 0.737 0.004 0.056 0.037 * CCL8 chemokine (C-C motif) ligand 8 0.724 0.002 0.036 0.025 * CCL18 chemokine (C-C motif) ligand 18 (pulmonary and activ 0.703 0.002 0.036 0.025 * CCL23 chemokine (C-C motif) ligand 23 0.701 0.002 0.036 0.025 * CCR6 chemokine (C-C motif) receptor 6 0.715 0.002 0.036 0.025 PTPN7 protein tyrosine phosphatase, non-receptor type 7 1.861 0.002 0.036 0.025 CD3Z CD3Z antigen, zeta polypeptide (TiT3 complex) 1.811 0.002 0.036 0.025 CD48 CD48 antigen (B-cell membrane protein) 1.809 0.002 0.036 0.025 IL10RA interleukin 10 receptor, alpha 1.806 0.002 0.036 0.025 SELL selectin L (lymphocyte adhesion molecule 1) 1.773 0.002 0.036 0.025 LCK lymphocyte-specific protein tyrosine kinase 1.745 0.002 0.036 0.025 -

Microenvironment-Derived ADAM28 Prevents Cancer Dissemination
www.oncotarget.com Oncotarget, 2018, Vol. 9, (No. 98), pp: 37185-37199 Research Paper Microenvironment-derived ADAM28 prevents cancer dissemination Catherine Gérard1, Céline Hubeau1, Oriane Carnet1, Marine Bellefroid1, Nor Eddine Sounni1, Silvia Blacher1, Guillaume Bendavid1,2, Markus Moser3, Reinhard Fässler3, Agnès Noel1, Didier Cataldo1,4,* and Natacha Rocks1,* 1Laboratory of Tumor and Development Biology, GIGA-Cancer and GIGA-I3, GIGA-Research, University of Liege, Liege, Belgium 2ENT Department, University Hospital of Liege, Liege, Belgium 3Max-Planck-Institute of Biochemistry, Department of Molecular Medicine, Martinsried, Germany 4Department of Respiratory Diseases, CHU Liege and University of Liege, Liege, Belgium *These authors have contributed equally to this work Correspondence to: Didier Cataldo, email: [email protected] Keywords: ADAM28; lung; metastasis; CD8+; T lymphocytes Received: July 27, 2018 Accepted: November 26, 2018 Published: December 14, 2018 Copyright: Gérard et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Previous studies have linked cancer cell-associated ADAM28 expression with tumor progression and metastatic dissemination. However, the role of host-derived ADAM28 in cancer dissemination processes remains unclear. Genetically engineered-mice fully deficient for ADAM28 unexpectedly display increased lung colonization by pulmonary, melanoma or breast tumor cells. In experimental tumor cell dissemination models, host ADAM28 deficiency is further associated with a decreased lung infiltration by CD8+ T lymphocytes. Notably, naive ADAM28-deficient mice already display a drastic reduction of CD8+ T cells in spleen which is further observed in lungs. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Novel Alternatively Spliced ADAM8 Isoforms Contribute to the Aggressive Bone Metastatic Phenotype of Lung Cancer
Oncogene (2010) 29, 3758–3769 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 www.nature.com/onc ORIGINAL ARTICLE Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer I Herna´ndez1, JL Moreno2, C Zandueta1, L Montuenga3,4 and F Lecanda1 1Adhesion and Metastasis Laboratory, Division of Oncology, University of Navarra, Pamplona, Spain; 2Department of Orthopaedics, University of Maryland School of Medicine, Baltimore, MD, USA; 3Department of Histology and Pathology, School of Medicine, University of Navarra, Pamplona, Spain and 4Biomarkers Laboratory, Center for Applied Biomedical Research (CIMA), University of Navarra, Pamplona, Spain ADAMs (a disintegrin and metalloprotease) are trans- survival rates for lung cancer are o15% in all developed membrane proteins involved in a variety of physiological countries. It is estimated that 30–40% of lung cancer processes and tumorigenesis. Recently, ADAM8 has been patients with advanced NSCLC suffer from bone associated with poor prognosis of lung cancer. However, metastasis (Coleman, 1997). Patients with bone metas- its contribution to tumorigenesis in the context of lung tasis experience pain, metabolic syndromes and spinal cancer metastasis remains unknown. Native ADAM8 cord compression associated with pathological fractures expression levels were lower in lung cancer cell lines. as a consequence of osteolytic lesions. In contrast, we identified and characterized two novel ADAMs (a disintegrin and metalloprotease) form a spliced isoforms encoding truncated proteins, D18a and large family of cell-surface proteins, which are char- D140, which were present in several tumor cell lines and not acterized by disintegrin and metalloproteinase domains, in normal cells. Overexpression of D18a protein resulted in that possess adhesive properties and proteolytic activ- enhanced invasive activity in vitro.ADAM8anditsD140 ities, respectively (Lu et al., 2007). -

Biochemical Characterization and Zinc Binding Group (Zbgs) Inhibition Studies on the Catalytic Domains of Mmp7 (Cdmmp7) and Mmp16 (Cdmmp16)
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Fan Meng Candidate for the Degree DOCTOR OF PHILOSOPHY ______________________________________ Director Dr. Michael W. Crowder ______________________________________ Dr. David L. Tierney ______________________________________ Dr. Carole Dabney-Smith ______________________________________ Dr. Christopher A. Makaroff ______________________________________ Graduate School Representative Dr. Hai-Fei Shi ABSTRACT BIOCHEMICAL CHARACTERIZATION AND ZINC BINDING GROUP (ZBGS) INHIBITION STUDIES ON THE CATALYTIC DOMAINS OF MMP7 (CDMMP7) AND MMP16 (CDMMP16) by Fan Meng Matrix metalloproteinase 7 (MMP7/matrilysin-1) and membrane type matrix metalloproteinase 16 (MMP16/MT3-MMP) have been implicated in the progression of pathological events, such as cancer and inflammatory diseases; therefore, these two MMPs are considered as viable drug targets. In this work, we (a) provide a review of the role(s) of MMPs in biology and of the previous efforts to target MMPs as therapeutics (Chapter 1), (b) describe our efforts at over-expression, purification, and characterization of the catalytic domains of MMP7 (cdMMP7) and MMP16 (cdMMP16) (Chapters 2 and 3), (c) present our efforts at the preparation and initial spectroscopic characterization of Co(II)-substituted analogs of cdMMP7 and cdMMP16 (Chapters 2 and 3), (d) present inhibition data on cdMMP7 and cdMMP16 using zinc binding groups (ZBG) as potential scaffolds for future inhibitors (Chapter 3), and (e) summarize our data in the context of previous results and suggest future directions (Chapter 4). The work described in this dissertation integrates biochemical (kinetic assays, inhibition studies, limited computational methods), spectroscopic (CD, UV-Vis, 1H-NMR, fluorescence, and EXAFS), and analytical (MALDI-TOF mass spectrometry, isothermal calorimetry) methods to provide a detailed structural and mechanistic view of these MMPs. -

ADAM23 Is Downregulated in Side Population and Suppresses Lung Metastasis of Lung Carcinoma Cells
ADAM23 is downregulated in side population and suppresses lung metastasis of lung carcinoma cells Masahide Ota,1,2 Satsuki Mochizuki,1 Masayuki Shimoda,1 Hitoshi Abe,1 Yuka Miyamae,1 Ken Ishii,3 Hiroshi Kimura2 and Yasunori Okada1,4 1Department of Pathology, Keio University School of Medicine, Tokyo; 2Second Department of Internal Medicine and Respiratory Medicine, Nara Medical University, Kashihara; 3Department of Orthopedic Surgery, Keio University School of Medicine, Tokyo; 4Department of Pathophysiology for Locomotive and Neoplastic Diseases, Juntendo University Graduate School of Medicine, Tokyo, Japan Key words Cancer cells contain a small population of cancer stem cells or cancer initiating ADAM23, colony formation, metastasis, non-small cell cells, which can be enriched in the side population (SP) after fluorescence acti- lung carcinoma cells, side population vated cell sorting. To examine the members of the ADAM, ADAMTS and MMP Correspondence gene families related to phenotypes of the SP and the main population (MP), we Yasunori Okada, Department of Pathophysiology for screened the expression of all the members in the propagated SP and MP of Locomotive and Neoplastic Diseases, Juntendo University A549 lung adenocarcinoma cells, and found that the relative expression ratio of Graduate School of Medicine, 2-1-1 Hongo, Bunkyo-ku, ADAM23 in the MP to the SP is most highly increased, but none of them are Tokyo 113-8421, Japan. increased in the SP. A similar result on the ADAM23 expression was obtained Tel: +81-3-5800-7531; Fax: +81-3-5800-7532; E-mail: [email protected] with another cell line, Calu-3 cells. Overexpression of ADAM23 inhibited colony formation, cell adhesion and migration, and knockdown of ADAM23 by shRNA Funding Information showed the reverse effects. -

VWF/ADAMTS13 Ratio As a Potential Biomarker for Early Detection Of
Takaya et al. BMC Gastroenterology (2019) 19:167 https://doi.org/10.1186/s12876-019-1082-1 RESEARCH ARTICLE Open Access VWF/ADAMTS13 ratio as a potential biomarker for early detection of hepatocellular carcinoma Hiroaki Takaya1, Tadashi Namisaki1* , Mitsuteru Kitade1, Kosuke Kaji1, Keisuke Nakanishi1, Yuki Tsuji1, Naotaka Shimozato1, Kei Moriya1, Kenichiro Seki1, Yasuhiko Sawada1, Soichiro Saikawa1, Shinya Sato1, Hideto Kawaratani1, Takemi Akahane1, Ryuichi Noguchi1, Masanori Matsumoto2 and Hitoshi Yoshiji1 Abstract Background: To investigate the von Willebrand factor to ADAMTS13 ratio as a potential biomarker for early detection of hepatocellular carcinoma (HCC) in cirrhosis. Methods: Serum levels of alpha-fetoprotein, des-γ-carboxy prothrombin, Lens culinaris agglutinin-reactive fraction of alpha-fetoprotein (alpha-fetoprotein-L3%), vascular endothelial growth factor, and vascular endothelial growth factor receptor-2, as well as the plasma levels of von Willebrand factor antigen (von Willebrand factor: Ag) and ADAMTS13 activity (ADAMTS13:AC), were evaluated in 41 cirrhotic patients with HCC undergoing radiofrequency ablation and in 20 cirrhotic patients without HCC. The diagnostic accuracy of each biomarker was evaluated using the receiver operating characteristic curve analysis. Results: The von Willebrand factor: Ag and von Willebrand factor: Ag/ADAMTS13:AC ratios were significantly higher in cirrhotic patients with HCC than in those without HCC (p <0.05andp < 0.01, respectively), whereas ADAMTS13:AC was significantly lower in those with HCC than those without HCC (p < 0.05). However, no relationship was observed between the von Willebrand factor: Ag/ADAMTS13:AC ratio and serum tumor markers such as alpha-fetoprotein, des-γ- carboxy prothrombin, and alpha-fetoprotein-L3%. Multivariate regression analysis identified von Willebrand factor: Ag/ ADAMTS13:AC ratio and alpha-fetoprotein-L3% as significant factors of HCC development. -

The Metalloproteinase ADAM28 Promotes Metabolic Dysfunction in Mice
International Journal of Molecular Sciences Article The Metalloproteinase ADAM28 Promotes Metabolic Dysfunction in Mice Lakshini Herat 1, Caroline Rudnicka 2, Yasunori Okada 3, Satsuki Mochizuki 4, Markus Schlaich 1,5 and Vance Matthews 1,* 1 Dobney Hypertension Centre, School of Medicine and Pharmacology, University of Western Australia, Crawley WA 6009, Australia; [email protected] (L.H.); [email protected] (M.S.) 2 Research Centre, Royal Perth Hospital, Perth WA 6000, Australia; [email protected] 3 Department of Pathophysiology for Locomotive and Neoplastic Diseases, Juntendo University Graduate School of Medicine, Tokyo 113-8421, Japan; [email protected] 4 Department of Surgery, National Defense Medical College, Saitama 359-8513, Japan; [email protected] 5 Department of Cardiology and Department of Nephrology, Royal Perth Hospital, Perth WA 6000, Australia * Correspondence: [email protected]; Tel.: +61-8-9224-0239; Fax: +61-8-9224-0374 Academic Editor: Masatoshi Maki Received: 17 February 2017; Accepted: 18 April 2017; Published: 21 April 2017 Abstract: Obesity and diabetes are major causes of morbidity and mortality globally. The current study builds upon our previous association studies highlighting that A Disintegrin And Metalloproteinase 28 (ADAM28) appears to be implicated in the pathogenesis of obesity and type 2 diabetes in humans. Our novel study characterised the expression of ADAM28 in mice with the metabolic syndrome and used molecular inhibition approaches to investigate the functional role of ADAM28 in the pathogenesis of high fat diet-induced obesity. We identified that ADAM28 mRNA and protein expression was markedly increased in the livers of mice with the metabolic syndrome. -

Expression Profiles and Clinical Correlations of Degradome Components in the Tumor Microenvironment of Head and Neck Squamous Cell Carcinoma
Published OnlineFirst March 21, 2010; DOI: 10.1158/1078-0432.CCR-09-2525 Clinical Human Cancer Biology Cancer Research Expression Profiles and Clinical Correlations of Degradome Components in the Tumor Microenvironment of Head and Neck Squamous Cell Carcinoma Angela Stokes1, Juho Joutsa6,7,8,9, Risto Ala-aho6,8, Mark Pitchers1, Caroline J. Pennington1, Craig Martin2, Don J. Premachandra3, Yasunori Okada10, Juha Peltonen4, Reidar Grénman5,7, Helen A. James1, Dylan R. Edwards1, and Veli-Matti Kähäri6,7,8 Abstract Purpose: Head and neck squamous cell carcinomas (HNSCC) are characterized by high morbidity and mortality, largely due to the high invasive and metastatic potential of these tumors, high recurrence rates, and low treatment responses. Proteinases have been implicated in several aspects of tumor growth and metastasis in a broad range of tumors including HNSCC. Experimental Design: Comprehensive expression profiling of proteinases [matrix metalloproteinases (MMPs), A disintegrin and metalloproteinase (ADAMs), and ADAMs with thrombospondin motif (ADAMTSs)] and their inhibitors [tissue inhibitor of metalloproteinases (TIMPs)] was done using quanti- tative real-time reverse transcription-PCR analysis of a large cohort of tissue samples representing the tumor (n = 83), the invasive margin (n = 41), and the adjacent tissue (n = 41) from 83 HNSCC patients, along with normal tissue controls (n = 13), as well as cell lines established from tumors of 34 HNSCC patients. Results: The results show specifically elevated gene expression of several proteinases, including MMP1, MMP3, MMP10, and MMP13 within tumor tissue and peritumoral adjacent tissue. In addition, the results identify several novel HNSCC-associated proteinases, including ADAM8, ADAM9, ADAM17, ADAM28, ADAMTS1, ADAMTS8, and ADAMTS15. -

ADAM28: Another Ambivalent Protease in Cancer
Cancer Letters 494 (2020) 18–26 Contents lists available at ScienceDirect Cancer Letters journal homepage: www.elsevier.com/locate/canlet ADAM28: Another ambivalent protease in cancer C´eline Hubeau a, Natacha Rocks b, Didier Cataldo a,c,* a Laboratory of Tumor and Development Biology, GIGA-Cancer, University of Li`ege, Li`ege, Belgium b Laboratory of Pharmaceutical Technology and Biopharmacy, CIRM, University of Li`ege, Li`ege, Belgium c Department of Respiratory Diseases, CHU of Li`ege, University of Li`ege, Li`ege, Belgium ARTICLE INFO ABSTRACT Keywords: Emergence of novel therapeutic options in a perspective of personalized therapy of cancer relies on the discovery ADAM28 of precise molecular mechanisms involved in the switch from a localized tumor to invasive metastasis spread. Cancer biomarker Pro-tumor functions have been mostly ascribed to proteolytic enzymes from the metalloproteinase family Cell proliferation including A Disintegrin And Metalloproteinases (ADAMs). Particularly, when expressed by cancer cells, ADAM28 Metastasis protease supports cancer cell proliferation, survival and migration as well as metastatic progression. In sharp Tumor microenvironment contrast, ADAM28 derived from the tumor microenvironment has shown to exert strong protective effects against deleterious metastasis dissemination. Indeed, depletion of host-derived ADAM28 (ADAM28 KO mice) accelerates colonization lung tissues, increases tumor foci implantation, and impairs T cell immune response. In this review, we outline specific ADAM28 functions when specifically expressed by carcinoma cells or by tumor microenvironment. Finally, we discuss about future research strategies that could be pursued to highlight new functions of this protease in cancer. 1. Introduction invasion of tumor cells as well as angiogenesis [3–5].