Isomerization of Allylic Alcohols to the Ketones Catalyzed by First-Row Transition Metal Pincer Complexes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alcohols Combined 1405
ALCOHOLS COMBINED 1405 Formulas: Table 1 MW: Table 1 CAS: Table 2 RTECS: Table 2 METHOD: 1405, Issue 1 EVALUATION: PARTIAL Issue 1: 15 March 2003 OSHA : Table 2 PROPERTIES: Table 1 NIOSH: Table 2 ACGIH: Table 2 COMPOUNDS: (1) n-butyl alcohol (4) n-propyl alcohol (7) cyclohexanol (2) sec-butyl alcohol (5) allyl alcohol (8) isoamyl alcohol (3) isobutyl alcohol (6) diacetone alcohol (9) methyl isobutyl carbinol SYNONYMS: See Table 3. SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPHY, FID (Coconut shell charcoal, 100 mg/50 mg) ANALYTE: Compounds above FLOW RATE: 0.01 to 0.2 L/min DESORPTION: 1 mL 5% 2-propanol in CS2 Compounds: (1-3 ) (4-9) VOL-MIN: 2 L 1 L INJECTION -MAX: 10 L 10 L VOLUME: 1 µL SHIPMENT: Routine TEMPERATURE -INJECTION: 220 °C SAMPLE -DETECTOR: 250 - 300 °C STABILITY: See Evaluation of Method. -COLUMN: 35 °C (7 minutes), to 60 °C at 5 °C/minute, hold 5 minutes, up to BLANKS: 2 to 10 field blanks per set 120 °C at 10 °C /minute, hold 3 minutes. CARRIER GAS: He, 4 mL/min ACCURACY COLUMN: Capillary, fused silica, 30 m x 0.32-mm RANGE STUDIED: Not studied [1, 2]. ID; 0.5 µm film polyethylene glycol, DB- wax or equivalent BIAS: Not determined CALIBRATION: Solutions of analyte in eluent (internal OVERALL standard optional) PRECISION (Ö ): Not determined rT RANGE: See EVALUATION OF METHOD. ACCURACY: Not determined ESTIMATED LOD: 1 µg each analyte per sample PRECISION: See EVALUATION OF METHOD. APPLICABILITY: This method may be used to determine two or more of the specified analytes simultaneously. -
Chapter 13.Pptx
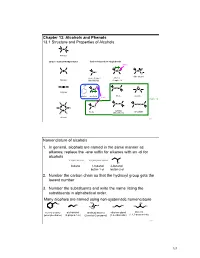
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

A New Approach to Prepare Polyethylene Glycol Allyl Glycidyl Ether
E3S Web of Conferences 267, 02004 (2021) https://doi.org/10.1051/e3sconf/202126702004 ICESCE 2021 A new approach to prepare Polyethylene Glycol Allyl Glycidyl Ether Huizhen Wang1*, Ruiyang Xie1, Mingjun Chen1*, Weihao Deng1, Kaixin Zhang2, Jiaqin Liu1 1School of Science, Xihua University, Chengdu 610039, China; 2Chengdu Jingyiqiang Environmental Protection Technology Co., Ltd. Abstract. The polyethylene glycol allyl glycidyl ether (PGAGE) is an important intermediate for preparing silicone softener that can be synthesized from allyl alcohol polyoxyethylene ether and epichlorohydrin (ECH). The performance parameters including the concentration of ECH, initial boron trifluoride diethyl etherate (BFEE) as well as CaCl2 quality were investigated respectively. The optimum process parameters which can get high capping and low by-product rate are as follows: the ECH concentration is 2.0 M, the initial BFEE concentration is 1.65mM, and the CaCl2 dosage is 1.65g/L. Under these conditions, the maximal yield can be improved to 91.36%, the percent of capping rate is higher than 98.16%, the residual concentration of F- is only 0.63 mg/L. concentrated basic solution, in which the total yield was between 90%~91% by Matsuoka et al. [10] also use the 1 Introduction two-step reaction to synthesize AGE based on the reaction Polyethylene glycol allyl glycidyl ether (PGAGE) and the of allyl alcohol with ECH using BFEE as the catalyst. allyl polyoxyethylene ether (APEG), tethering with both Their results demonstrated that the yield reaches 82% alkene and epoxy groups, are widely used as fabric under the following condition: n (ECH) : n (allyl alcohol): finishing agent [1-2] , reactive diluent [3] , cross-linking (catalysis) = 1: (1~3) : (0.01~0.002). -

Title Selective Reduction of Acrolein to Allyl Alcohol by a Vapor Phase
Selective Reduction of Acrolein to Allyl Alcohol by a Vapor Title Phase Hydrogen Transfer Reaction Author(s) Kunichika, Sango; Oka, Shinzaburo; Sakai, Mutsuji Bulletin of the Institute for Chemical Research, Kyoto Citation University (1966), 44(3): 215-220 Issue Date 1966-10-31 URL http://hdl.handle.net/2433/76125 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University Selective Reduction of Acrolein to Allyl Alcohol by a Vapor Phase Hydrogen Transfer Reaction Sango KUNICHIKA,Shinzaburo OKA and Mutsuji SAKAI* (KunichikaLaboratory) (ReceivedJune 15, 1966) Selectivereduction of acroleinto ally! alcoholhas been achievedby a vapor phase hydrogen transferreaction using isopropyl alcohol as a hydrogendonor. It was foundthat the preferredcatalyst was 50 weight% zinc oxide-magnesiumoxide catalyst prepared by calcininga mixtureof zincnitrate and magnesiumoxide at 500°C. The optimumconditions were found to be as follows;reaction tem- perature,near 200°C,total feed rate, 45 moles/1.of catalyst/hr.,feed ratio of isopropylalcohol per acrolein,10 moles. Under theseconditions, the yieldsof allyl alcoholof 80-65% at 80-90% conversion of acroleinwere obtained. INTRODUCTION Acrolein has two reactive functional groups, one is carbonyl group and the other is carbon-carbon double bond. Selective reduction of acrolein to allyl alcohol has been studied by several investigators. The reaction was generally carried out by the following two methods: (1) the Meerwein-Ponndorf-Verley reduction1-3), (2) vapor phase hydrogen transfer reaction with various metal oxide catalysts using an alcohol as a hydrogen donor4-11> The liquid phase reaction has some technical difficulties owing to the polymeri- zation of acrolein especially at high conversion. Therefore, in the present work, the more promising vapor phase reaction has been studied to find an effective catalyst and favorable reaction conditions. -

Conversion of Glycerol Into Allyl Alcohol Over Potassium-Supported Zirconia-Iron Oxide Catalyst
Title Conversion of glycerol into allyl alcohol over potassium-supported zirconia-iron oxide catalyst Author(s) Konaka, Aya; Tago, Teruoki; Yoshikawa, Takuya; Nakamura, Ayaka; Masuda, Takao Applied catalysis b-environmental, 146, 267-273 Citation https://doi.org/10.1016/j.apcatb.2013.03.007 Issue Date 2014-03 Doc URL http://hdl.handle.net/2115/54715 Type article (author version) File Information Manuscript_Glycerol to Allyl Alcohol .pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Conversion of Glycerol into Allyl Alcohol over Potassium-Supported Zirconia-Iron Oxide Catalyst Aya Konaka, Teruoki Tago*, Takuya Yoshikawa, Ayaka Nakamura, and Takao Masuda Research Group of Chemical Engineering, Division of Chemical Process Engineering, Faculty of Engineering, Hokkaido University, N13W8, Kita-ku, Sapporo, Hokkaido, Japan 060-8628 * Corresponding author should be addressed E-mail: [email protected] Tel: +81-11-706-6551 Fax: +81-11-706-6552 1 Abstract The catalytic conversion of glycerol was performed with iron oxide-based catalysts for production of allyl alcohol using a fixed-bed flow reactor at 623 K under atmospheric pressure. The glycerol dehydration proceeds on acid sites of catalysts while the allyl alcohol production is assumed to be catalyzed by non-acidic sites of catalysts through a hydrogen transfer mechanism. Different alkali metals, including Na, K, Rb, and Cs were supported on ZrO2-FeOX and all of them gave impressively higher allyl alcohol yield and suppressed glycerol dehydration due to the reduced catalyst acidic property. K-supported ZrO2-FeOX (K/ZrO2-FeOX) was chosen for further studies, and allyl alcohol yield remarkably increased up to 27 mol%-C at the K content of 3-5 mol%. -

United States Patent (19) (11) Patent Number: 4,968,451 Scheibel Et Al
United States Patent (19) (11) Patent Number: 4,968,451 Scheibel et al. 45) Date of Patent: Nov. 6, 1990 (54) SOIL RELEASE AGENTS HAVING ALLYL-DERVED SULFONATED END CAPS FOREIGN PATENT DOCUMENTS 155710 9/1985 European Pat. Off. (75) Inventors: Jeffrey J. Scheibel; Eugene P. 180356 5/1986 European Pat. Off. Gosselink, both of Cincinnati, Ohio 1475798 6/1977 United Kingdom . (73) Assignee: The Procter & Gamble Company, OTHER PUBLICATIONS Cincinnati, Ohio S. C. Bright, C. E. Stubbs and L. Thompson, J. Appl. (21) Appl. No.: 474,709 Chem. Biotechnol, 1975, vol. 25, pp. 901-912. 22) Filed: Jan. 29, 1990 Norton et al., J. Org. Chem, vol. 33, No. 11, pp. 4158-4165 (1967). Related U.S. Application Data Primary Examiner-A. Lionel Clingman (63) Continuation of Ser. No. 237,598, Aug. 26, 1988, aban Attorney, Agent, or Firm-Leonard W. Lewis; Jerry J. doned. Yetter (51 Int, C. .................... CO7C 309/05; C08G 63/68; 57 ABSTRACT C11D 3/37 (52) U.S. C. ............................... 252/549; 252/174.19; The present invention relates to novel soil release 252/539; 252/558; 252/DIG. 15 agents, which are particular sulfonated linear tere (58) Field of Search ......................................... 252/549 phthalate ester oligomers (S.T.E. oligomers). The S.T.E. oligomers are especially suitable for formulation (56 References Cited into laundry products such as laundry detergents or U.S. PATENT DOCUMENTS fabric conditioners. Thus formulated, they provide ef. 3,821,169 6/1974 Duddey et al. ..................... 528/293 fective soil release treatments for fabrics laundered in 4,156,073 5/1979 Login ............. -

Selective Liquid-Phase Oxidation of Allyl Alcohol to Glycidol Over MWW Type Titanosilicalite
Reac Kinet Mech Cat (2011) 103:451–462 DOI 10.1007/s11144-011-0312-5 Selective liquid-phase oxidation of allyl alcohol to glycidol over MWW type titanosilicalite Anna Fajdek • Agnieszka Wro´blewska • Eugeniusz Milchert Received: 4 February 2011 / Accepted: 23 March 2011 / Published online: 7 April 2011 Ó The Author(s) 2011. This article is published with open access at Springerlink.com Abstract The results of the epoxidation of allyl alcohol with 30% hydrogen peroxide over the Ti-MWW catalyst have been presented. The studies were carried out under atmospheric pressure and in the presence of methanol as a solvent. The influence of the following technological parameters on the course of epoxidation was examined: temperature (20–60 °C), the molar ratio of AA/H2O2 (1:1–5:1), methanol concentration (5–90 wt%), catalyst content (0–5.0 wt%), reaction time (5–300 min) and intensity of stirring (0–500 rpm). Keywords Glycidol Á Ti-MWW catalyst Á Liquid-phase epoxidation Á Hydrogen peroxide Introduction Zeolites are crystalline aluminosilicates with 133 known frameworks. Their pores are highly monodisperse and have a fixed directionality within the crystal. The pores in these systems often have a character of cages and connection tubes. The uniformity of the channels in the zeolitic system means that they are extremely selective adsorbents for small molecules of particular geometries and sizes and so make highly selective catalytic supports and adsorbents. Due to the pore sizes comparable to the molecular dimensions, zeolites are also called molecular sieves [1]. MWW (MCM-22) is a molecular sieve which crystallizes as thin sheets or plates and has a unique and unusual crystal structure. -

Estimation of the Allyl Group in Starch Derivatives
Estimation of the allyl group in starch derivatives K. TIHLÁRIK, V. KÉRY, and M. PAŠTEKA Institute of Chemistry, Centre for Chemical Research, Slovak Academy of Sciences, CS-84238 Bratislava Received 7 June 1988 For estimation of the allyl groups in allylstarch a method which involves addition of mercury (II) acetate to double bonds and the subsequent acid- imetric titration has been applied. The method was verified by the allyl alcohol estimation and the results obtained in the estimation of two allyl starch samples were compared with the values of elemental analyses and with the degree of substitution values calculated from these analyses. There was a good agreement between both approaches. The method is especially suitable for the derivatives with low degree of substitution. The results have been evaluated statistically. Для определения аллильных групп в аллилкрахмале был применен метод присоединения уксуснокислой ртути по двойным связям с по следующим ацидиметрическим титрованием. Метод был проверен путем определения количества аллилового спирта, и результаты, полу ченные при анализе двух образцов аллилкрахмала, были сопоставлены с результатами элементарного анализа этих образцов и с величинами степени замещения, рассчитанными исходя из результатов элементар ного анализа. Было обнаружено хорошее совпадение. Данный метод особенно пригоден для производных с низкой степенью замещения. Полученные результаты были статистически обработаны. Estimation of the double bonds is of importance in the products of petrol and food industries, in pharmacy, and is also important in polysaccharide deriva tives, e.g. in allylcellulose and allylstarch. The latter derivatives are applied not only for technical purposes but serve as intermediates, mainly in the controlled polymerization, for the production of mixed polymers for special purposes. -

Allyl Alcohol Product Safety Bulletin
Allyl Alcohol Product Safety Bulletin lyondellbasell.com Foreword Lyondell Chemical Company and Lyondell Chemie Nederland Telephone numbers for transport emergencies: BV (“Lyondell”), LyondellBasell companies, are dedicated to continuous improvement in product, health, safety and environmental CHEMTREC 1-800-424-9300 performance. Included in this effort is a commitment to support our International (call collect) 202-483-7616 customers by providing guidance and information on the safe use of our products. For Lyondell, environmentally sound operations, like CANUTEC (in Canada) 1-613-996-6666 environmentally sound products, make good business sense. SETIQ (in the Mexican Republic) 01-800-00-214-00 Lyondell Product Safety Bulletins are prepared by our Product (Calls originating in Mexico City or the Metropolitan Area) +5-559-1588 Stewardship Team. The data reflect the best information available (Calls originating elsewhere) +52-555-559-1588 from public and industry sources. This document is provided to support the safe handling, use, storage, transportation and ultimate LyondellBasell TDI disposal of our chemical products. (Transportation Distribution Incident) This Product Safety Bulletin should be evaluated to determine reporting Hotline applicability of your specific requirements. The government regulations, industry standards cited in this bulletin are primarily 1-800-245-4532 (North America) applicable within the United States. Please make sure you review +32-3-575-1235 (Europe) the corresponding government regulations, industry standards and guidelines for your specific country or region as that might have an Contact information for additional product impact on your operations. information: Lyondell’s ready to support our customers’ safe use of our products. LyondellBasell Customer Service For additional information and assistance, please contact your +1-888-777-0232 (North America) Lyondell customer representative. -

Dissociation Constants of Organic Acids and Bases
DISSOCIATION CONSTANTS OF ORGANIC ACIDS AND BASES This table lists the dissociation (ionization) constants of over pKa + pKb = pKwater = 14.00 (at 25°C) 1070 organic acids, bases, and amphoteric compounds. All data apply to dilute aqueous solutions and are presented as values of Compounds are listed by molecular formula in Hill order. pKa, which is defined as the negative of the logarithm of the equi- librium constant K for the reaction a References HA H+ + A- 1. Perrin, D. D., Dissociation Constants of Organic Bases in Aqueous i.e., Solution, Butterworths, London, 1965; Supplement, 1972. 2. Serjeant, E. P., and Dempsey, B., Ionization Constants of Organic Acids + - Ka = [H ][A ]/[HA] in Aqueous Solution, Pergamon, Oxford, 1979. 3. Albert, A., “Ionization Constants of Heterocyclic Substances”, in where [H+], etc. represent the concentrations of the respective Katritzky, A. R., Ed., Physical Methods in Heterocyclic Chemistry, - species in mol/L. It follows that pKa = pH + log[HA] – log[A ], so Academic Press, New York, 1963. 4. Sober, H.A., Ed., CRC Handbook of Biochemistry, CRC Press, Boca that a solution with 50% dissociation has pH equal to the pKa of the acid. Raton, FL, 1968. 5. Perrin, D. D., Dempsey, B., and Serjeant, E. P., pK Prediction for Data for bases are presented as pK values for the conjugate acid, a a Organic Acids and Bases, Chapman and Hall, London, 1981. i.e., for the reaction 6. Albert, A., and Serjeant, E. P., The Determination of Ionization + + Constants, Third Edition, Chapman and Hall, London, 1984. BH H + B 7. Budavari, S., Ed., The Merck Index, Twelth Edition, Merck & Co., Whitehouse Station, NJ, 1996. -

Chemical Compatibility Chart
Chemical Compatibility Chart 1 Inorganic Acids 1 2 Organic acids X 2 3 Caustics X X 3 4 Amines & Alkanolamines X X 4 5 Halogenated Compounds X X X 5 6 Alcohols, Glycols & Glycol Ethers X 6 7 Aldehydes X X X X X 7 8 Ketone X X X X 8 9 Saturated Hydrocarbons 9 10 Aromatic Hydrocarbons X 10 11 Olefins X X 11 12 Petrolum Oils 12 13 Esters X X X 13 14 Monomers & Polymerizable Esters X X X X X X 14 15 Phenols X X X X 15 16 Alkylene Oxides X X X X X X X X 16 17 Cyanohydrins X X X X X X X 17 18 Nitriles X X X X X 18 19 Ammonia X X X X X X X X X 19 20 Halogens X X X X X X X X X X X X 20 21 Ethers X X X 21 22 Phosphorus, Elemental X X X X 22 23 Sulfur, Molten X X X X X X 23 24 Acid Anhydrides X X X X X X X X X X 24 X Represents Unsafe Combinations Represents Safe Combinations Group 1: Inorganic Acids Dichloropropane Chlorosulfonic acid Dichloropropene Hydrochloric acid (aqueous) Ethyl chloride Hydrofluoric acid (aqueous) Ethylene dibromide Hydrogen chloride (anhydrous) Ethylene dichloride Hydrogen fluoride (anhydrous) Methyl bromide Nitric acid Methyl chloride Oleum Methylene chloride Phosphoric acid Monochlorodifluoromethane Sulfuric acid Perchloroethylene Propylene dichloride Group 2: Organic Acids 1,2,4-Trichlorobenzene Acetic acid 1,1,1-Trichloroethane Butyric acid (n-) Trichloroethylene Formic acid Trichlorofluoromethane Propionic acid Rosin Oil Group 6: Alcohols, Glycols and Glycol Ethers Tall oil Allyl alcohol Amyl alcohol Group 3: Caustics 1,4-Butanediol Caustic potash solution Butyl alcohol (iso, n, sec, tert) Caustic soda solution Butylene -

United States Patent to (Ii) Patent Number: 4,956,076 Awbrey (45) Date of Patent: Sep
United States Patent to (ii) Patent Number: 4,956,076 Awbrey (45) Date of Patent: Sep. 11, 1990 (54) METHOD OF SCAVENGING HYDROGEN 2,993,862 7/1961 Monroe et al. ..................... 252/396 2,993,863 7/1961 Monroe et al. ...... ... 252/396 HALIDES FROM LIQUID 3,310,497 3/1964 Hudson et al. .. ... 252/396 HYDROCARBONACEOUSMEDIUMS 3,375,460 5/1965 Barbie ......... 208/262.1 75 Inventor: Spencer S. Awbrey, Spring, Tex. 3,400,171 9/1968 Van Pool ......................... 208/262.1 3,403,198 9/1968 Van Pool ......................... 208/262.1 (73) Assignee: Betz Laboratories, Inc., Trevose, Pa. 3,413,237 11/1968 Foroulis .............................. 252/.393 9/1973 Sun et al. .... 208/262.1 3,761,534 21 Appl. No.: 414,049 3,779,905 12/1973 Stedman ............................... 208/47 (22 Filed: Sep. 28, 1989 4,430,196 2/1984 Niu ........................................ 208/47 (51) Int. Cl................................................. C10G 7/10 Primary Examiner-Helane E. Myers (52) U.S.C. .................................... 208/291; 208/474; Attorney, Agent, or Firm-Alexander D. Ricci; Bruce E. 208/262.1; 208/262.5; 585/950; 585/864; Peacock 252/346; 210/109; 210/705; 210/723; 210/729 ABSTRACT (58 Field of Search ............... 585/950; 208/47, 262.1, 57 208/291,47; 252/396; 210/705, 723, 729 Methods of scavenging hydrogen halide species from hydrocarbon liquids are disclosed. Preferred scavengers (56) References Cited includes allyl alcohol, benzyl alcohol and tertiary alco U.S. PATENT DOCUMENTS hols such as tert-butyl alcohol. 2,415,161 2/1947 Camp .................................... 208/47 2,603,622 7/1952 Berger et al.