2020.03.30.015305V1.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio In
microorganisms Review The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease Spase Stojanov 1,2, Aleš Berlec 1,2 and Borut Štrukelj 1,2,* 1 Faculty of Pharmacy, University of Ljubljana, SI-1000 Ljubljana, Slovenia; [email protected] (S.S.); [email protected] (A.B.) 2 Department of Biotechnology, Jožef Stefan Institute, SI-1000 Ljubljana, Slovenia * Correspondence: borut.strukelj@ffa.uni-lj.si Received: 16 September 2020; Accepted: 31 October 2020; Published: 1 November 2020 Abstract: The two most important bacterial phyla in the gastrointestinal tract, Firmicutes and Bacteroidetes, have gained much attention in recent years. The Firmicutes/Bacteroidetes (F/B) ratio is widely accepted to have an important influence in maintaining normal intestinal homeostasis. Increased or decreased F/B ratio is regarded as dysbiosis, whereby the former is usually observed with obesity, and the latter with inflammatory bowel disease (IBD). Probiotics as live microorganisms can confer health benefits to the host when administered in adequate amounts. There is considerable evidence of their nutritional and immunosuppressive properties including reports that elucidate the association of probiotics with the F/B ratio, obesity, and IBD. Orally administered probiotics can contribute to the restoration of dysbiotic microbiota and to the prevention of obesity or IBD. However, as the effects of different probiotics on the F/B ratio differ, selecting the appropriate species or mixture is crucial. The most commonly tested probiotics for modifying the F/B ratio and treating obesity and IBD are from the genus Lactobacillus. In this paper, we review the effects of probiotics on the F/B ratio that lead to weight loss or immunosuppression. -

Evaluation of 16S Rrna Primer Sets for Characterisation of Microbiota in Paediatric Patients with Autism Spectrum Disorder L
www.nature.com/scientificreports OPEN Evaluation of 16S rRNA primer sets for characterisation of microbiota in paediatric patients with autism spectrum disorder L. Palkova1,2, A. Tomova3, G. Repiska3, K. Babinska3, B. Bokor4, I. Mikula5, G. Minarik2, D. Ostatnikova3 & K. Soltys4,6* Abstract intestinal microbiota is becoming a signifcant marker that refects diferences between health and disease status also in terms of gut-brain axis communication. Studies show that children with autism spectrum disorder (ASD) often have a mix of gut microbes that is distinct from the neurotypical children. Various assays are being used for microbiota investigation and were considered to be universal. However, newer studies showed that protocol for preparing DNA sequencing libraries is a key factor infuencing results of microbiota investigation. The choice of DNA amplifcation primers seems to be the crucial for the outcome of analysis. In our study, we have tested 3 primer sets to investigate diferences in outcome of sequencing analysis of microbiota in children with ASD. We found out that primers detected diferent portion of bacteria in samples especially at phylum level; signifcantly higher abundance of Bacteroides and lower Firmicutes were detected using 515f/806r compared to 27f/1492r and 27f*/1495f primers. So, the question is whether a gold standard of Firmicutes/Bacteroidetes ratio is a valuable and reliable universal marker, since two primer sets towards 16S rRNA can provide opposite information. Moreover, signifcantly higher relative abundance of Proteobacteria was detected using 27f/1492r. The beta diversity of sample groups difered remarkably and so the number of observed bacterial genera. An advent of massively parallel sequencing (MPS) technologies has revolutionized an investigating of human microbiota1. -

Appendix III: OTU's Found to Be Differentially Abundant Between CD and Control Patients Via Metagenomeseq Analysis
Appendix III: OTU's found to be differentially abundant between CD and control patients via metagenomeSeq analysis OTU Log2 (FC CD / FDR Adjusted Phylum Class Order Family Genus Species Number Control) p value 518372 Firmicutes Clostridia Clostridiales Ruminococcaceae Faecalibacterium prausnitzii 2.16 5.69E-08 194497 Firmicutes Clostridia Clostridiales Ruminococcaceae NA NA 2.15 8.93E-06 175761 Firmicutes Clostridia Clostridiales Ruminococcaceae NA NA 5.74 1.99E-05 193709 Firmicutes Clostridia Clostridiales Ruminococcaceae NA NA 2.40 2.14E-05 4464079 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides NA 7.79 0.000123188 20421 Firmicutes Clostridia Clostridiales Lachnospiraceae Coprococcus NA 1.19 0.00013719 3044876 Firmicutes Clostridia Clostridiales Lachnospiraceae [Ruminococcus] gnavus -4.32 0.000194983 184000 Firmicutes Clostridia Clostridiales Ruminococcaceae Faecalibacterium prausnitzii 2.81 0.000306032 4392484 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides NA 5.53 0.000339948 528715 Firmicutes Clostridia Clostridiales Ruminococcaceae Faecalibacterium prausnitzii 2.17 0.000722263 186707 Firmicutes Clostridia Clostridiales NA NA NA 2.28 0.001028539 193101 Firmicutes Clostridia Clostridiales Ruminococcaceae NA NA 1.90 0.001230738 339685 Firmicutes Clostridia Clostridiales Peptococcaceae Peptococcus NA 3.52 0.001382447 101237 Firmicutes Clostridia Clostridiales NA NA NA 2.64 0.001415109 347690 Firmicutes Clostridia Clostridiales Ruminococcaceae Oscillospira NA 3.18 0.00152075 2110315 Firmicutes Clostridia -

Xiexin Tang Improves the Symptom of Type 2 Diabetic Rats by Modulation of the Gut Microbiota
www.nature.com/scientificreports OPEN Xiexin Tang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota Received: 30 August 2017 Xiaoyan Wei, Jinhua Tao , Suwei Xiao, Shu Jiang, Erxin Shang, Zhenhua Zhu, Dawei Qian Accepted: 13 February 2018 & Jinao Duan Published: xx xx xxxx Type 2 diabetes mellitus (T2DM), a chronic metabolic disease which severely impairs peoples’ quality of life, currently attracted worldwide concerns. There are growing evidences that gut microbiota can exert a great impact on the development of T2DM. Xiexin Tang (XXT), a traditional Chinese medicine prescription, has been clinically used to treat diabetes for thousands of years. However, few researches are investigated on the modulation of gut microbiota community by XXT which will be very helpful to unravel how it works. In this study, bacterial communities were analyzed based on high-throughput 16S rRNA gene sequencing. Results indicated that XXT could notably shape the gut microbiota. T2DM rats treated with XXT exhibited obvious changes in the composition of the gut microbiota, especially for some short chain fatty acids producing and anti-infammatory bacteria such as Adlercreutzia, Alloprevotella, Barnesiella, [Eubacterium] Ventriosum group, Blautia, Lachnospiraceae UCG-001, Papillibacter and Prevotellaceae NK3B31 group. Additionally, XXT could also signifcantly ameliorate hyperglycemia, lipid metabolism dysfunction and infammation in T2DM rats. Moreover, the correlation analysis illustrated that the key microbiota had a close relationship with the T2DM related indexes. The results probably provided useful information for further investigation on its active mechanism and clinical application. T2DM, a chronic metabolic disease characterized by hyperglycemia as a result of insufcient insulin secretion, insulin action or both1, is estimated that its numbers in the adults will increase by 55% by 20352. -

Development of the Equine Hindgut Microbiome in Semi-Feral and Domestic Conventionally-Managed Foals Meredith K
Tavenner et al. Animal Microbiome (2020) 2:43 Animal Microbiome https://doi.org/10.1186/s42523-020-00060-6 RESEARCH ARTICLE Open Access Development of the equine hindgut microbiome in semi-feral and domestic conventionally-managed foals Meredith K. Tavenner1, Sue M. McDonnell2 and Amy S. Biddle1* Abstract Background: Early development of the gut microbiome is an essential part of neonate health in animals. It is unclear whether the acquisition of gut microbes is different between domesticated animals and their wild counterparts. In this study, fecal samples from ten domestic conventionally managed (DCM) Standardbred and ten semi-feral managed (SFM) Shetland-type pony foals and dams were compared using 16S rRNA sequencing to identify differences in the development of the foal hindgut microbiome related to time and management. Results: Gut microbiome diversity of dams was lower than foals overall and within groups, and foals from both groups at Week 1 had less diverse gut microbiomes than subsequent weeks. The core microbiomes of SFM dams and foals had more taxa overall, and greater numbers of taxa within species groups when compared to DCM dams and foals. The gut microbiomes of SFM foals demonstrated enhanced diversity of key groups: Verrucomicrobia (RFP12), Ruminococcaceae, Fusobacterium spp., and Bacteroides spp., based on age and management. Lactic acid bacteria Lactobacillus spp. and other Lactobacillaceae genera were enriched only in DCM foals, specifically during their second and third week of life. Predicted microbiome functions estimated computationally suggested that SFM foals had higher mean sequence counts for taxa contributing to the digestion of lipids, simple and complex carbohydrates, and protein. -

WO 2018/064165 A2 (.Pdf)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/064165 A2 05 April 2018 (05.04.2018) W !P O PCT (51) International Patent Classification: Published: A61K 35/74 (20 15.0 1) C12N 1/21 (2006 .01) — without international search report and to be republished (21) International Application Number: upon receipt of that report (Rule 48.2(g)) PCT/US2017/053717 — with sequence listing part of description (Rule 5.2(a)) (22) International Filing Date: 27 September 2017 (27.09.2017) (25) Filing Language: English (26) Publication Langi English (30) Priority Data: 62/400,372 27 September 2016 (27.09.2016) US 62/508,885 19 May 2017 (19.05.2017) US 62/557,566 12 September 2017 (12.09.2017) US (71) Applicant: BOARD OF REGENTS, THE UNIVERSI¬ TY OF TEXAS SYSTEM [US/US]; 210 West 7th St., Austin, TX 78701 (US). (72) Inventors: WARGO, Jennifer; 1814 Bissonnet St., Hous ton, TX 77005 (US). GOPALAKRISHNAN, Vanch- eswaran; 7900 Cambridge, Apt. 10-lb, Houston, TX 77054 (US). (74) Agent: BYRD, Marshall, P.; Parker Highlander PLLC, 1120 S. Capital Of Texas Highway, Bldg. One, Suite 200, Austin, TX 78746 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

Download (PDF)
Supplemental Table 1. OTU (Operational Taxonomic Unit) level taxonomic analysis of gut microbiota Case-control Average ± STDEV (%) Wilcoxon OTU Phylum Class Order Family Genus Patient (n = 15) Control (n = 16) p OTU_366 Firmicutes Clostridia Clostridiales Ruminococcaceae Oscillospira 0.0189 ± 0.0217 0.00174 ± 0.00429 0.004** OTU_155 Firmicutes Clostridia Clostridiales Ruminococcaceae Ruminococcus 0.0554 ± 0.0769 0.0161 ± 0.0461 0.006** OTU_352 Firmicutes Clostridia Clostridiales Ruminococcaceae Ruminococcus 0.0119 ± 0.0111 0.00227 ± 0.00413 0.007** OTU_719 Firmicutes Clostridia Clostridiales Ruminococcaceae Oscillospira 0.00150 ± 0.00190 0.000159 ± 0.000341 0.007** OTU_620 Firmicutes Clostridia Clostridiales Ruminococcaceae Unclassified 0.00573 ± 0.0115 0.0000333 ± 0.000129 0.008** OTU_47 Firmicutes Clostridia Clostridiales Lachnospiraceae Lachnospira 1.44 ± 2.04 0.183 ± 0.337 0.011* OTU_769 Firmicutes Clostridia Clostridiales Ruminococcaceae Unclassified 0.000857 ± 0.00175 0 0.014* OTU_747 Firmicutes Clostridia Clostridiales Ruminococcaceae Anaerotruncus 0.000771 ± 0.00190 0 0.014* OTU_748 Firmicutes Clostridia Clostridiales Ruminococcaceae Unclassified 0.0135 ± 0.0141 0.00266 ± 0.00420 0.015* OTU_81 Bacteroidetes Bacteroidia Bacteroidales [Barnesiellaceae] Unclassified 0.434 ± 0.436 0.0708 ± 0.126 0.017* OTU_214 Firmicutes Clostridia Clostridiales Ruminococcaceae Unclassified 0.0254 ± 0.0368 0.00496 ± 0.00959 0.020* OTU_113 Actinobacteria Coriobacteriia Coriobacteriales Coriobacteriaceae Unclassified 0 0.189 ± 0.363 0.021* OTU_100 Bacteroidetes -

Extensive Microbial Diversity Within the Chicken Gut Microbiome Revealed by Metagenomics and Culture
Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture Rachel Gilroy1, Anuradha Ravi1, Maria Getino2, Isabella Pursley2, Daniel L. Horton2, Nabil-Fareed Alikhan1, Dave Baker1, Karim Gharbi3, Neil Hall3,4, Mick Watson5, Evelien M. Adriaenssens1, Ebenezer Foster-Nyarko1, Sheikh Jarju6, Arss Secka7, Martin Antonio6, Aharon Oren8, Roy R. Chaudhuri9, Roberto La Ragione2, Falk Hildebrand1,3 and Mark J. Pallen1,2,4 1 Quadram Institute Bioscience, Norwich, UK 2 School of Veterinary Medicine, University of Surrey, Guildford, UK 3 Earlham Institute, Norwich Research Park, Norwich, UK 4 University of East Anglia, Norwich, UK 5 Roslin Institute, University of Edinburgh, Edinburgh, UK 6 Medical Research Council Unit The Gambia at the London School of Hygiene and Tropical Medicine, Atlantic Boulevard, Banjul, The Gambia 7 West Africa Livestock Innovation Centre, Banjul, The Gambia 8 Department of Plant and Environmental Sciences, The Alexander Silberman Institute of Life Sciences, Edmond J. Safra Campus, Hebrew University of Jerusalem, Jerusalem, Israel 9 Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, UK ABSTRACT Background: The chicken is the most abundant food animal in the world. However, despite its importance, the chicken gut microbiome remains largely undefined. Here, we exploit culture-independent and culture-dependent approaches to reveal extensive taxonomic diversity within this complex microbial community. Results: We performed metagenomic sequencing of fifty chicken faecal samples from Submitted 4 December 2020 two breeds and analysed these, alongside all (n = 582) relevant publicly available Accepted 22 January 2021 chicken metagenomes, to cluster over 20 million non-redundant genes and to Published 6 April 2021 construct over 5,500 metagenome-assembled bacterial genomes. -

Gut Microbiota Features Associated with Clostridioides Difficile Colonization in Dairy Calves
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Gut microbiota features associated with Clostridioides 2 difficile colonization in dairy calves 3 4 Laurel E. Redding1, Alexander S. Berry2,3, Nagaraju Indugu1, Elizabeth Huang1, Daniel P. Beiting3, Dipti 5 Pitta1 6 7 8 1Department of Clinical Sciences, University of Pennsylvania, School of Veterinary Medicine, Kennett 9 Square, PA, USA 10 11 2Division of Gastroenterology, Hepatology, and Nutrition, Children’s Hospital of Philadelphia, Philadelphia, 12 PA, USA 13 14 3Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, 15 USA 16 17 18 Corresponding author: Laurel Redding, [email protected] 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Diarrheal disease, a major cause of morbidity and mortality in dairy calves, is strongly associated with the 22 health and composition of the gut microbiome. Clostridioides difficile is an opportunistic pathogen that 23 proliferates and can produce enterotoxins when the host experiences gut dysbiosis. -

Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease
biomedicines Article Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease Ashani Lecamwasam 1,2,3,*, Tiffanie M. Nelson 4, Leni Rivera 3, Elif I. Ekinci 2,5, Richard Saffery 1,6 and Karen M. Dwyer 3 1 Epigenetics Research, Murdoch Children’s Research Institute, VIC 3052, Australia; [email protected] 2 Department of Endocrinology, Austin Health, VIC 3079, Australia; [email protected] 3 School of Medicine, Faculty of Health, Deakin University, VIC 3220, Australia; [email protected] (L.R.); [email protected] (K.M.D.) 4 Menzies Health Institute Queensland, Griffith University, QLD 4222, Australia; [email protected] 5 Department of Medicine, University of Melbourne, VIC 3010, Australia 6 Department of Paediatrics, University of Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +613-8341-6200; Fax: +613-9348-1391 Abstract: (1) Background: Individuals with diabetes and chronic kidney disease display gut dysbiosis when compared to healthy controls. However, it is unknown whether there is a change in dysbiosis across the stages of diabetic chronic kidney disease. We investigated a cross-sectional study of patients with early and late diabetes associated chronic kidney disease to identify possible microbial differences between these two groups and across each of the stages of diabetic chronic kidney disease. (2) Methods: This cross-sectional study recruited 95 adults. DNA extracted from collected stool samples were used for 16S rRNA sequencing to identify the bacterial community in the Citation: Lecamwasam, A.; Nelson, gut. (3) Results: The phylum Firmicutes was the most abundant and its mean relative abundance T.M.; Rivera, L.; Ekinci, E.I.; Saffery, was similar in the early and late chronic kidney disease group, 45.99 ± 0.58% and 49.39 ± 0.55%, R.; Dwyer, K.M. -
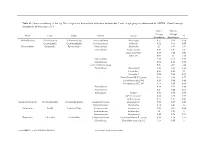
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58